Dermatomyositis and Undifferentiated Nasopharyngeal Carcinoma. A Rare Presentation of a Rare Malignancy
Maria Belen Mateo1, Giancarlo Candela1, Maria Jesus Delgado1, Ricardo Hitt2, María Jose Echarri2
2.Department of Oncology, Hospital Universitario Severo Ochoa, Leganes, Spain.
Citation : Mateo MB, Candela G, Delgado MJ, Hitt R, Echarri MJ. Dermatomyositis and Undifferentiated Nasopharyngeal Carcinoma. A Rare Presentation of a Rare Malignancy. Asclepius Med Case Rep 2018;1(2):1-4.
The incidence of nasopharyngeal carcinoma (NPC) is 0.5-25 cases over 100.000 of all malignancies. Undifferentiated NPC type is the most frequent of all in endemic areas. Dermatomyositis (DM) is a relatively rare disorder, associated with malignancy in 10-50% of cases. Few cases have been reported by the literature referring to DM and undifferentiated NPC in Caucasian population [1]. We present a 65-year-old man with a history of relapsing bilateral serous mucoid otitis, who developed skin lesions and muscle fatigability, compatible with DM as a paraneoplastic syndrome (PNS). He received standard therapy for NPC and 4 months after the end of treatment; we observed a complete remission of both the tumor and PNS. The existence of DM must aware physicians of underlying cancer, due to the fact that it is frequently associated with a large number of tumors. However, PNS and NPC are an unusual presentation.
Dermatomyositis, paraneoplastic syndrome, undifferentiated nasopharyngeal carcinoma
INTRODUCTION
Paraneoplastic dermatomyositis associated with Undifferentiated nasopharyngeal carcinoma is uncommon [1]. The purpose of this case is to reflect the importance of searching for a neoplasm when DM appears in adulthood and to expose a rare presentation a NPC.
CASE REPORT
In September 2016, a 65-year-old Caucasian man visited his general practitioner after having noticed sudden dermatological abnormalities, affecting his face, arms, hands, chest, and upper back: An itchy erythematous rash, which he related to sun exposure. He was diagnosed with subacute eczema and followed treatment with topic steroids and antihistaminic per os, without any improvement.
His previous medical history noted: Severe smoker, usual snoring (no sleeping test), and hypertension. He had a bilateral serous mucoid otitis (SMO) having failed medical treatment, since 2014; with a normal fibroscopyin January 2016. A right and left myringotomy and transtympanic drainage had been performed during the same month the skin lesions appeared.
Cutaneous lesions persisted, and in January 2017, he developed diplopia, dysphagia, ageusia, and major snoring. He consulted the otorhinolaryngologist, who noticed a VI left cranial nerve palsy and submaxilar lymphadenopathies. A new fibroscopy was made, revealing a nasopharyngeal mass. Imaging (cerebral and cervical magnetic resonance imaging and body computered tomography) and mass biopsy revealed an undifferentiated nasopharyngeal carcinoma (NPC), VEB+, and stage T4N2cM0.
He was referred to the oncology department; anamnesis discovered, besides, he had started with proximal muscle lower limb weakness at the same time skin lesion appeared. Physical exam showed facial and scalp erythema, exanthematic erythema on the upper chest and less evident on the upper back (V sign), elbow and knuckle erythema (Gottron's sign) [Figures 1-3]. Strength on all limbs was 5/5, no muscle atrophy was noticed. Further physical examination was normal.
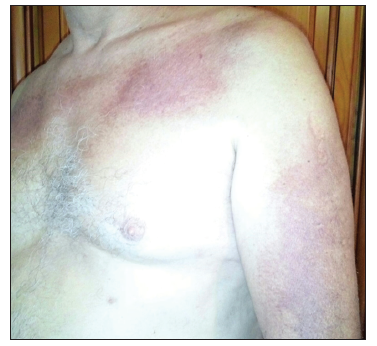
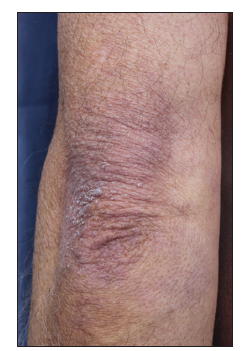
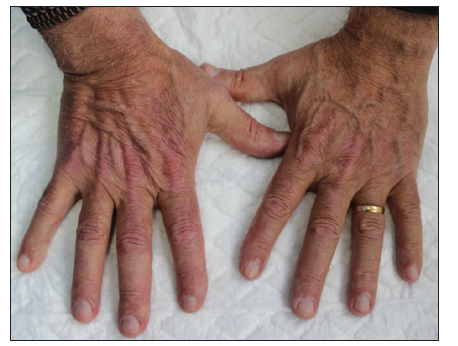
Blood tests exposed aldolase at 17 U/L (normal: 0-8), with normal creatinine kinase, lactate dehydrogenase (LDH), glutamic oxaloacetic transaminase (GOT), and glutamic pyruvic transaminase (GPT); and erythrocyte sedimentation rate (ESR) at 22 mm1h (normal: 0-10). Rheumatoid factor, crioaglutinines, antinuclear antibodies, antimitochondrial antibodies, ENAs (SSA, SSB, RNP/Sm, Scl70, and antiJo1), and antisynthetase antibodies (PL-7, PL-12, signal recognition particle [SRP]- 54, Mi-2, Ku, Pm-Scl, Scl- 70) resulted negative. Acetylcholine receptor antibodies were positive. Electromyography noted deltoid damage compatible with myopathy. Deltoid muscle histopathological findings (perifascial atrophy and lymphocytaryperimysial infiltrate) confirmed dermatomyositis (DM) [Figure 4].
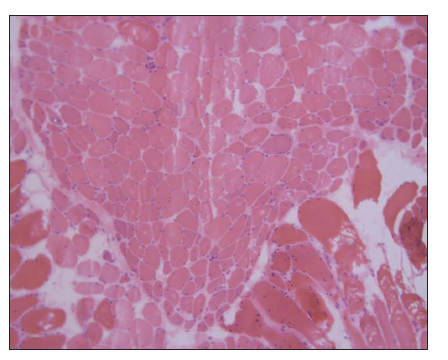
Treatment consisted of RTOG standard protocol: Cisplatin 100 mg/m2 day 1-22-43 plus intensity-modulated radiotherapy. After chemoradiotherapy, the patient presented intense mucositis and xerostomia. Moreover, during this time, dysphagia appeared and required hospitalization for enteral nutrition. An esophageal manometry was performed, showing upper and medium esophageal dysmotility. Transthoracic echocardiogram dismissed cardiac damage. He started oral prednisone 10 mg/day for the dysphagia and continued maintenance with a low dose of corticoids until withdrawal. Meanwhile, he completed treatment with 3 cycles of Cisplatin plus 5 fluorouracil according to protocol. 1 year after having completed chemoradiation therapy, neither cutaneous lesions, muscle weakness, nor dysphagia persisted, and complete response of the tumor was achieved.
DISCUSSION
The incidence of NPC is 0.5-25 cases over 100.000 of all malignancies [1] It is classified into three different subtypes (WHO): Type 1, differentiated; type 2, poorly differentiated; and type 3, undifferentiated. Undifferentiated NPC is the most frequent in endemic areas (63% of the total NPC: Asia and North Africa), and it is usually related to Ebstein Barr virus infection.
DM is a relatively rare disorder, with an incidence varying between 2 and 19 cases per 1 million population. DM is a systemic inflammatory disorder. Bohan and Dakalas suggested the use of five criteria to define DM: (1) Progressive and symmetrical proximal muscle weakness of proximal limb muscle and anterior neck flexors, with or without dysphagia, or respiratory muscle damage: (2) Typical dermatological signs: Heliotrope rash, Gottron's sign, periorbitary edema, and erythematous rash: (3) Elevated muscle enzyme levels (creatine phosphokinase, aldolase, LDH, GOT, and GPT): (4) Electromyography compatible with myopathy and: (5) Muscle biopsy compatible with myositis. Defining as definitive DM the presence of rash plus 3 or 4 criteria. Nevertheless, cutaneous damage in paraneoplastic DM is unspecific, and it frequently appears in an amyopathic form [2].
It was first described by Stretz as a paraneoplastic syndrome (PS) in 1916. It is associated with malignancy in 10-50% of cases. The risk of having cancer in DM patients is 6 to 7 times higher than the normal population. Juvenile DM is rarely associated with a neoplastic origin [3]. By contrast, if DM presents itself in adulthood, cancer must be thoroughly seeked for. DM can appear before, simultaneously with, or after a cancer diagnosis. The highest incidence of cancer in DM appears during the 1st year since the onset and reduces after the first 5 years. DM has been reported in ovarian, breast, lung, pancreas, and gastrointestinal cancers in Europe and America; and NPC in East Asia. The association DM with undifferentiated NPC in the Caucasian population is exceptional, with only three cases published [4-6].
Pathogenesis of DM as PNS is unknown. It is believed that association between DM and PNS may be due to the expression of autoantigens common to both the tumoral tissue and myopathic muscular tissue. The immune systemic response to the tumoral tissue may affect muscular tissue. There are three types of specific antibodies associated with DM: Antisynthetase, anti-SRP (anti-SRP) antibodies, and anti-Mi2. Moreover, there have been described cancerassociated antibodies which attack the transcription intermediary factor-1 gamma (anti-p155 and anti-p155/140) and the nuclear matrix protein (NPX)-2 (anti-MJ or anti-p140). Meanwhile, the presence of myositis-specific antibodies appears to be associated with a decreased risk of malignancy [2,7,8].
Some risk factors for PNS are mentioned in the literature, such as older age at disease onset, dysphagia, and resistance to treatment; and some biopsy findings (cutaneous necrosis, capillary damage, and leukocytoclastic vasculitis). However, a lower risk of DM PNS is described when there is pulmonary damage and when anti-MDA5antibodies are expressed [8,9]. It has not been depicted major severity of DM when paraneoplastic. Despite this, an apparition or outbreak of DM should make us suspicious of possible tumor relapse.
The treatment of paraneoplastic DM consists of treating the neoplasm. Nevertheless, it is normally observed a poor response to usual treatment. Oral corticosteroids may be required. When no response is achieved, immunosuppression with methotrexate, azathioprine, or cyclosporine may be effective. High dose of intravenous immunoglobulins has also been used, especially for treating pruritus.
Our patient had a 2-year evolution SMO, with normal endoscopical results. DM signs, standing for erythema, and Gottron sign started 3 months before cancer diagnosis were achieved. Probably the absence of heliotrope rash delayed malignancy's diagnosis. He had also muscle and esophageal damage, without pulmonary affection. In our case, it existed and elevation of aldolase levels, with normal results not only for the rest of muscle enzymes determinations but also for specific myositis antibodies. We did not test cancer-related antibodies due to the fact that diagnosis could be established with Bohan's criteria. Acetylcholine receptor antibodies were initially determined when diplopia was noticed. Regardless of the positive result compatible with seropositive Myasthenia gravis, tumor extension explained diplopia, and muscle affection was compatible with DM. We have presented this clinical case because of the low incidence of undifferentiated NPC on the Caucasian population, the usual delay of the diagnosis, and it's an exceptional association with DM [10,11].
CONCLUSION
In cases of pneumocephalus where the exact cause is not well documented, an extensive investigation is recommended to ascertain the etiology before the institution of hyperbaric oxygen therapy.
References
- Mahdavifar N, Towhidi F, Makhsosi BR, Pakzad R, Moini A, Ahmadi A, et al. Incidence and mortality of nasopharynx cancer and its relationship with human development index in the world in 2012. World J Oncol 2016;7:109-18.
- Requena C, Alfaro A, Traves V, Nagore E, Llombart B, Serra C, et al. Paraneoplastic Dermatomyositis. Report of 12 cases. Actas Dermosifiliogr 2014;105:675-82.
- Morris P, Dare J. Juvenile dermatomyositis as a paraneoplastic phenomenon: An update. J Pediatr Hematol Oncol 2010;32:189-91.
- Chakroun A, Guigay J, Lusinchi A, Marandas P, Janot F, Hartl DM, et al. Paraneoplastic dermatomyositis accompanying nasopharyngeal carcinoma: Diagnosis, treatment and prognosis. Eur Ann Otorhinolaryngol Head Neck Dis 2011;128:127-31.
- Ziani FZ, Brahmi SA, Najib R, Kanab R, Arifi S, Mernissi FZ, et al. Paraneoplastic dermatomyositis revealing an undifferentiated nasopharyngeal carcinoma: About a case. Pan Afr Med J 2016;24:29.
- Botsios C, Ostuni P, Boscolo-Rizzo P, Da Mosto MC, Punzi L, Marchiori C, et al. Dermatomyositis and malignancy of the pharynx in caucasian patients: Report of two observations. Rheumatol Int 2003;23:309-11.
- Dourmishev LA. Inflammatory myopathies with cutaneous involvement: From diagnosis to therapy. Folia Med (Plovdiv) 2017;59:7-13.
- Chinoy H, Fertig N, Oddis CV, Ollier WE, Cooper RG. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis 2007;66:1345-9.
- Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola- Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): A retrospective study. J Am Acad Dermatol 2011;65:25-34.
- Hill EK, King PH, Hughey LC. Dermatomyositis and concomitant overlap myasthenic syndrome: A rare presentation. J Am Acad Dermatol 2011;65:e150-2.
- Santos E, Coutinho E, Martins da Silva A, Marinho A, Vasconcelos C, Taipa R, et al. Inflammatory myopathy associated with myasthenia gravis with and without thymic pathology: Report of four cases and literature review.Autoimmun Rev 2017;16:644-9.