Inherited Cerebral Small Vessel Disease, Vascular Cognitive Impairment, Classical Cardiovascular Risk Factors, and Preventive Measures - Lessons from Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
George P. Paraskevas1, Vasilios C. Constantinides1, Panagiotis G. Paraskevas2, Elisabeth Kapaki1
2.Department of Nursing, Technological Educational Institute of Crete, School of Health and Welfare Services, Heraklion, Stauromenos 71004 Crete, Greece.
Citation : GP, Constantinides VC, Paraskevas PG, Kapaki E. Inherited Cerebral Small Vessel disease, Vascular Cognitive Impairment, Classical Cardiovascular Risk Factors, and Preventive Measures - Lessons from Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leucoencephalopathy. Asclepius Med Res Rev 2018;1(1):1-6.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is caused by mutations in NOTCH 3 gene, and it is the most common cause of inherited cerebral small vessel disease and vascular cognitive impairment (VCI). There are manynew insights on the mechanisms of cognitive decline, relevant to the evolution of VCI of any cause. The most important issue is that, although being a genetic disease, the phenotype may be worsened by classical cardiovascular risk factors, especially hypertension and smoking. Control of these risk factors may delay disease progression and disability in CADASIL and, currently, is strongly recommended.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy, diabetes, dyslipidemia, hypertension, smoking, lacunar stroke, microbleeds, vascular cognitive impairment, white matter hyperintensities
INTRODUCTION
Vascular cognitive impairment (VCI) is the second most common cause of cognitive impairment in the elderly,[1] and among many mechanisms and causes, subcortical small vessel disease (SSVD) is probably the most frequent [2]. It is usually a sporadic disorder of the elderly, due to classical cardiovascular risk factors, including hypertension diabetes and dyslipidemia [3]. Studies on VCI may recruit such patients, and this is frequently done. However, up to two-third of such elderly patients may harbor concomitant Alzheimer's disease pathology,[4] which may contribute significantly to symptoms and the degree of cognitive impairment, and thus, they suffer from mixed rather than pure VCI [5]. The use of biomarkers, including cerebrospinal fluid biomarkers for Alzheimer's disease, may help in the correct patient selection [2,3] However, such diagnostic procedures are costly and not available in all clinical settings. On the other hand, SSVD due to inherited causes, although much rarer, may offer not only animal models[6] but also homogeneous patient samples, usually with no additional pathology, more suitable for studying pure VCI and understanding the mechanisms of and relationship between SSVD, lacunar stroke, and VCI [7].
Inherited cerebral small vessel diseases comprise a group of rare monogenic disorders leading to cerebrovascular disease and stroke [8]. Among these, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is considered to be the most common (or least rare) monogenic cause of inherited stroke, subcortical vascular disease, and vascular dementia,[9] comprising ~60% of genetic microischemic leukoencephalopathies [10]. It is due to mutations in the NOTCH3 gene at chromosmome 19q12,[11] which causes alterations in vessel wall of arterioles (including deposition of granular osmiophilic material), resulting in brain tissue ischemia [12].
Typically, migraine (usually with aura) at about the age of 30 or early ischemic events (transient ischemic attacks and lacunar stroke) at 41-50 years are the presenting symptoms [13] Neuroimaging features include multiple and, later on, confluent ischemic lesions in the white matter (WM) and basal ganglia with characteristic involvement of the anterior temporal WM and external capsule [14]. As the disease progresses and ischemic lesion load increases, behavioralpsychiatric manifestations and cognitive decline become evident as well as bilateral pyramidal and pseudobulbar signs lead to vascular dementia, significant motor disability, and premature death usually at or before 65-70 years [15]. A significant variation in phenotypic presentation, disease severity, and rate of deterioration exists among different families carrying the same mutation and even among patients of the same family. Thus, patients with normalappearing magnetic resonance imaging at the 4th decade of life,[16] with a later age of disease onset, [17] with a later onset of stroke,[18] sometimes as late as the 8th decade,[19] or with oligosymptomatic presentations[20] are increasingly being recognized. On the other hand, rapid deterioration with severe psychiatric symptoms and dementia at the 4th-5th decades is not uncommon [21].
CORRELATES OF DEMENTIA SEVERITY
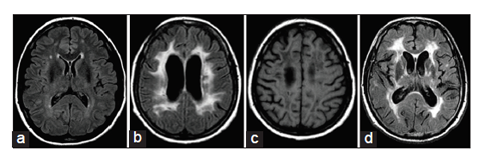
Very early in the disease process, when patients are still asymptomatic or have only migraine and show no evidence of cognitive impairment, MRIs may initially seem normal. However, deposition of protein material in the lymphatic drainage pathways in the walls of cerebral vasculature may impair drainage of interstitial fluid [22]. Venous vasculature is also decreased,[23] while decreased contrast between gray and WM in T1 images has been reported,[24] possibly related to signal alterations in normal appearing WM. Alterations in WM that is normal appearing in conventional images have been shown by calculating magnetization transfer values [25] and may be observed by diffusion tensor imaging [26]. As decreased drainage of interstitial fluid becomes more severe and, additionally, some degree of ischemia is present, WM (probably intramyelinic) edema occurs [27] and WM hyperintensities (WMH) appear [Figure 1a] and become progressively numerous. The load of WMH alone showed a negative correlation with global cognitive measures, frontosubcortical and/or executive function tests, verbal fluency, and delayed memory scores [28]. However, despite executive and attentional deficits, patients at this stage usually show no more than mild disability and no frank dementia [28].
Dilated perivascular spaces increase with age in the entire brain, and it has been suggested that dilated spaces located in temporal lobes and subinsular areas are strongly and specifically associated with WMH, while dilated spaces in the WM independently correlate with cognitive decline [29]. Microbleeds in deep or cortical locations may also be present and they are independently associated with executive and frontosubcortical dysfunction [28]. Emotional symptoms of any type (depressive or non-depressive) seem to be associated with microbleeds in thalamus and cortex [30]. However, WMH alone, even in the presence of microbleeds, produces subclinical or mild cognitive impairment, usually reaching no more than the threshold of mild dementia [28]. Significant axonal damage in addition to demyelination [31] and, especially, lacunes [32] [ Figure 1b and c] is usually required for more severe cognitive dysfunction, since such lesions are more able to interrupt important cortico-cortical and corticosubcortical circuits and produce disconnection syndromes.
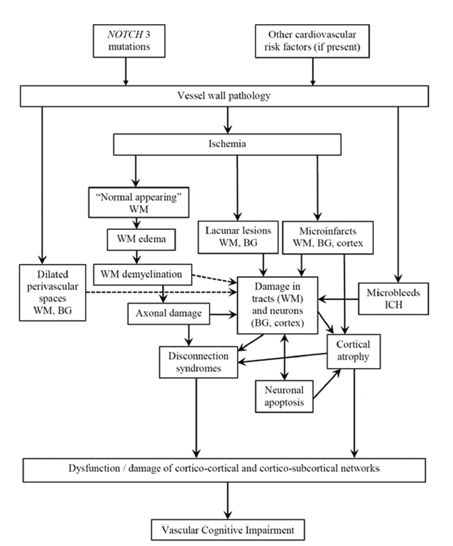
It seems that the type and extent of lesions are not only the parameters affecting the severity of cognitive impairment but also their "strategic" location [Figure 2].
Arteriolar pathology, WM pathology, and axonal abnormalities, although widespread, may severely affect the WM of the frontal lobe [Figure 1d], affecting frontosubcortical and/or frontocortical networks [31]. Based on neuroimaging, five areas show the maximum influence in processing speed: lacunar load in the left anterior thalamic radiation and left cingulum and WMH in the left forceps minor, left parahippocampal WM and left corticospinal tract [33]. Since these areas are related with important frontosubcortical neuronal circuits (especially, dorsolateral prefrontal and cingulate circuits), it is not surprising that the above lesions in these areas roughly explain one-third of the total variance in processing speed [33]. In addition, lacunar lesion load in anterior thalamic radiation is associated with the reduced thickness of medial frontal cortex (which, in turn, is associated with deficits in processing speed) and reduced thickness of right occipitotemporal cortex, the latter (lingual, fusiform, and parahippocampal cortices) also being associated with abnormal processing speed [34]. It has been suggested that, within the WM of the frontal lobe, the superior longitudinal fasciculus may be affected even earlier than the cingulum bundle [31]. This may result in a frontal disconnection syndrome that gradually involves the parietal and temporal lobes.
CAUSES OF CLINICAL VARIABILITY AND CORRELATES OF ISCHEMIC LESION LOAD
Although it is generally accepted that most of the different NOTCH3 mutations have little effect in phenotypic variability, a minority of mutations affecting the NOTCH3 ligand binding domain (EGFR10-11) may be associated with the decreased load of WMH and better cognitive function [35]. Multiple variants of other genes, each with small effect, may also influence the load of WM lesions, partly explaining some of the phenotypic variations among patients [36] Sex affects clinical presentation. In males, migraine onset occurs 6 years later, and the first lacunar stroke occurs 7 years earlier than in females [13]. Unfortunately, the above factors are non-modifiable.
It is long known that, in the appropriate clinical setting, the absence of cardiovascular risk factors increases the diagnostic probability of CADASIL. This should not be interpreted that patients with CADASIL have no cardiovascular risk factors. The presence of hypertension, diabetes, dyslipidemia, and thrombophilia has been reported in many patients and should not preclude the diagnosis of CADASIL [37,38]. If present, such factors may modify the clinical features [38]. Hypertension independently increases the risk for stroke [38-40] and disability due to dementia [41]. Smoking also increases the risk of stroke [38,42] and dementia [42]. Hypertension [40] and diabetes with increased HbA1c may increase the risk for microbleeds [40,43].
The above observations lead to suggestions for controlling these risk factors, especially hypertension and smoking, in an attempt to delay lacunar stroke, disease progress, and functional disability [38, 42] Such a disease modifying approach is being followed during the last decade and is currently recommended [44].
Recently, it has been shown that the epidemiology of CADASIL is changing [18]. The median age of first stroke is higher than previously estimated, especially in men. Men diagnosed after 2006 experiences their first stroke at a median age of 56 and 10 years later than those diagnosed before 2006.[18] Furthermore, in patients over 58 years old, 38% remain independent,[18] as compared to 14% of patients over 60 years, before 2000 [45]. Such a favorable change in the natural history of CADASIL may be partly due to a better knowledge of the disorder, resulting in increased suspicion, better diagnosis, and identification of more "benign" cases. However, control of risk factors may have also contributed. Indeed, in two monozygotic twins with CADASIL, the one that followed preventive measures such as physical activity and early control of dyslipidemia with statin showed less severe imaging findings and experienced his first stroke 14 years later than the one which was smoker and delayed controlling dyslipidemia [46].
CONCLUSIONS
Despite being a genetic disease, the phenotype of CADASIL may be worsened by classical cardiovascular risk factors. Control of such risk factors may delay disease progression and disability and, currently, should be strongly recommended.
References
- Wallin A, Roman GC, Esiri M, Kettunen P, Svensson J, Paraskevas GP, et al. Update on vascular cognitive impairment associated with subcortical small-vessel disease. J Alzheimers Dis 2018;62:1417-144.
- Wallin A, Kapaki E, Boban M, Engelborghs S, Hermann DM, Huisa B, et al. Biochemical markers in vascular cognitive impairment associated with subcortical small vessel disease - A consensus report. BMC Neurol 2017;17:102.
- Rosenberg GA, Wallin A, Wardlaw JM, Markus HS, Montaner J, Wolfson L, et al. Consensus statement for diagnosis of subcortical small vessel disease. J Cereb Blood Flow Metab 2016;36:6-25.
- Attems J, Jellinger KA. The overlap between vascular disease and Alzheimer's disease-lessons from pathology. BMC Med 2014;12:206.
- Kalaria RN. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer's disease. Acta Neuropathol 2016;131:659-85.
- Joutel A, Faraci FM. Cerebral small vessel disease: Insights and opportunities from mouse models of collagen IV-related small vessel disease and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 2014;45:1215-21.
- Caplan LR. Lacunar infarction and small vessel disease: Pathology and pathophysiology. J Stroke 2015;17:2-6.
- Choi JC. Genetics of cerebral small vessel disease. J Stroke 2015;17:7-16.
- Tan RY, Markus HS. Monogenic causes of stroke: Now and the future. J Neurol 2015;262:2601-16.
- Ayrignac X, Carra-Dalliere C, de Champfleur NM, Denier C, Aubourg P, Bellesme C, et al. Adult-onset genetic leukoencephalopathies: A MRI pattern-based approach in a comprehensive study of 154 patients. Brain 2015;138:284-92.
- Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 1996;383:707-10.
- Tikka S, Baumann M, Siitonen M, Pasanen P, Poyhonen M, Myllykangas L, et al. CADASIL and CARASIL. Brain Pathol 2014;24:525-44.
- Tan RY, Markus HS. CADASIL: Migraine, encephalopathy, stroke and their inter-relationships. PLoS One 2016;11:e0157613.
- Singhal S, Rich P,Markus HS. The spatial distribution of MR imaging abnormalities in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy and their relationship to age and clinical features. Am J Neuroradiol 2005;26:2481-7.
- Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. CADASIL. Lancet Neurol 2009;8:643-53.
- Samoes R, Alves JE, Taipa R, Silva J, Pires MM, Pereira-Monteiro JM. CADASIL: MRI maybe normal in the fourthdecade of life-a case report. Cephalalgia 2016;36:1082-5.
- Liao YC, Hsiao CT, Fuh JL, Chern CM, Lee WJ, Guo YC, et al. Characterization of CADASIL among the han Chinese in Taiwan: Distinct genotypic and phenotypic profiles. PLoS One 2015;10:e0136501.
- Moreton FC, Razvi SS, Davidson R, Muir KW. Changingclinical patterns and increasingprevalence in CADASIL. Acta Neurol Scand 2014;130:197-203.
- Pescini F, Bianchi S, Salvadori E, Poggesi A, Dotti MT, Federico A, et al. A pathogenic mutation on exon 21 of the NOTCH3 genecausing CADASIL in an octogenarian paucisymptomatic patient. J Neurol Sci 2008;267:170-3.
- Paraskevas GP, Constantinides VC, Yapijakis C, Kararizou E, Kapaki EN, Bougea A. Recognition of cerebral autosomal dominant Arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) in two oligosymptomatic sisters with low CADASIL scale scores and a venousdysplasia: Report of a novel Greek family. J Stroke Cerebrovasc Dis 2018;27:e191-5.
- Paraskevas GP, Constantinides VC, Bougea A, Gerakoulis E, Yapijakis C, Kararizou E, et al. Cerebralautosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy presenting with postpartum psychosis and late-onset stroke. Future Neurol 2016;1:207-13.
- Weller RO, Hawkes CA, Kalaria RN, Werring DJ, Carare RO. White matter changes in dementia: Role of impaired drainage of interstitialfluid. Brain Pathol 2015;25:63-78.
- De Guio F, Vignaud A, Ropele S, Duering M, Duchesnay E, Chabriat H, et al. Loss of venousintegrity in cerebral small vessel disease: A 7-T MRI study in cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Stroke 2014;45:2124-6.
- De Guio F, Reyes S, Duering M, Pirpamer L, Chabriat H, Jouvent E. Decreased T1 contrast between gray matter and normal-appearing white matter in CADASIL. Am J Neuroradiol 2014;35:72-6.
- Iannucci G, Dichgans M, Rovaris M, Bruning R, Gasser T, Giacomotti L, et al. Correlations between clinical findings and magnetization transfer imaging metrics of tissue damage in individuals with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 2001;32:643-8.
- Chabriat H, Pappata S, Poupon C, Clark CA, Vahedi K, Poupon F, et al. Clinicalseverity in CADASIL related to ultrastructural damage in white matter: In vivo study with diffusion tensor MRI. Stroke 1999;30:2637-43.
- De Guio F, Mangin JF, Duering M, Ropele S, Chabriat H, Jouvent E. White matteredema at the early stage of cerebralautosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 2015;46:258-61.
- Benisty S, Reyes S, Godin O, Herve D, Zieren N, Jouvent E, et al. White-matter lesions without lacunar infarcts in CADASIL. J Alzheimer's Dis 2012;29:903-11.
- Yao M, Herve D, Jouvent E, Duering M, Reyes S, Godin O,et al. Dilated perivascular spaces in small-vesseldisease: A study in CADASIL. Cerebrovasc Dis 2014;37:155-63.
- Noh SM, Chung SJ, Kim KK, Kang DW, Lim YM, Kwon SU, et al. Emotional disturbance in CADASIL: Its impact on quality of life and caregiverburden. Cerebrovasc Dis 2014;37:188-94.
- Craggs LJ, Yamamoto Y, Ihara M, Fenwick R, Burke M, Oakley AE, et al. White matter pathology and disconnection in the frontal lobe in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Neuropathol Appl Neurobiol 2014;40:591-602.
- Viswanathan A, Gschwendtner A, Guichard JP, Buffon F, Cumurciuc R, O'Sullivan M, et al. Lacunar lesions are independently associated with disability and cognitiveimpairment in CADASIL. Neurology 2007;69:172-9.
- Duering M, Gonik M, Malik R, Zieren N, Reyes S, Jouvent E, et al. Identification of a strategicbrain network underlying processing speed deficits in vascular cognitive impairment. Neuroimage 2013;66:177-83.
- Righart R, Duering M, Gonik M, Jouvent E, Reyes S, Herve D, et al. Impact of regional cortical and subcortical changes on processing speed in cerebral small vessel disease. Neuroimage Clin 2013;2:854-61.
- Monet-Lepretre M, Bardot B, Lemaire B, Domenga V, Godin O, Dichgans M, et al. Distinct phenotypic and functional features of CADASIL mutations in the Notch3 ligand binding domain. Brain 2009;132:1601-12.
- Opherk C, Gonik M, Duering M, Malik R, Jouvent E, Herve D, et al. Genome-wide genotyping demonstrates a polygenic risk score associated with white matter hyperintensity volume in CADASIL. Stroke 2014;45:968-72.
- Pantoni L, Sarti C, Pescini F, Bianchi S, Bartolini L, Nencini P, et al. Thrombophilic risk factors and unusual clinical features in three Italian CADASIL patients. Eur J Neurol 2004;11:782-7.
- Adib-Samii P, Brice G, Martin RJ, Markus HS. Clinical spectrum of CADASIL and the effect of cardiovascular risk factors on phenotype. Study in 200 consecutively recruited individuals. Stroke 2010;41:630-4.
- Ling Y, De Guio F, Duering M, Jouvent E, Herve D, Godin O, et al. Predictors and clinical impact of incident lacunes in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 2017;48:283-9.
- Lee JS, Kang CH, Park SQ, Choi HA, Sim KB. Clinical significance of cerebral microbleeds locations in CADASIL with R544C NOTCH3 mutation. PLoS One 2015;10:e0118163.
- Ciolli L, Pescini F, Salvadori E, Del Bene A, Pracucci G, Poggesi A, et al. Influence of vascular risk factors and neuropsychological profile on functional performances in CADASIL: Results from the microvascular leukoencephalopathy study (MILES). Eur J Neurol 2014;21:65-71.
- Chabriat H, Herve D, Duering M, Godin O, Jouvent E, Opherk C, et al. Predictors of Clinical worsening in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: Prospective cohort study. Stroke 2016;47:4-11.
- Viswanathan A, Guichard JP, Gschwendtner A, Buffon F, Cumurcuic R, Boutron C, et al. Blood pressure and haemoglobin A1c are associated with microhaemorrhage in CADASIL: A two-centre cohort study. Brain 2006;129:2375-83.
- Di Donato I, Bianchi S, De Stefano N, Dichgans M, Dotti MT, Duering M, et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)as a model of small vessel disease: Update on clinical, diagnostic, and management aspects. BMC Med 2017;15:41.
- Dichgans M, Mayer M, Uttner I, Bruning R, Muller-Hocker J, Rungger G, et al. The phenotypic spectrum of CADASIL: Clinical findings in 102 cases. Ann Neurol 1998;44:731-9.
- Mykkanen K, Junna M, Amberla K, Bronge L, Kaariainen H, Poyhonen M, et al. Different clinical phenotypes in monozygotic CADASIL twins with a novel NOTCH3 mutation. Stroke 2009;40:2215-8.