The Main Molecular Bases of Amyotrophic Lateral Sclerosis: An Integrating Review
Leonardo Ricardo Pizani1, Patricia Ucelli Simioni1,2
2.Institute of Biosciences, Universidade Estadual Paulista, UNESP, Rio Claro, SP, Brazil
Citation : Pizani LR, Simioni PU. The Main Molecular Bases of Amyotrophic Lateral Sclerosis: An Integrating Review.Asclepius Med Res Rev 2018;1(1):1-8.
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disease that affects the lower and upper motor neurons. Its etiology is unknown, despite several studies that point to risk factors and genetic for the disease. It does not have a definitive treatment and the diagnosis, most of the time, is time consuming and demands varied criteria for adequate definition and classification. In the search for new treatments, gene therapy with gene insertion aims to reduce the impact of ALS on the patient. The study of molecular basis as the root cause of the disease becomes necessary to advance the discovery of effective treatment and a rapid diagnosis, aiming at improving the quality of life of the ALS patients. Therefore, the present work had the objective to bring an update of the literature on ALS, as well as to describe the main molecular bases involved in the causative/ causative process of this disease.
Amyotrophic lateral sclerosis, motor neurons, nervous system diseases.
BACKGROUND
Vmyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of the lower motor nervous system (LMNS) and upper motor nervous System(UMNS), which affects the cerebral cortex, brain stem, and spinal cord, being classified according to the initially compromised neurological region [1]. Depending on the degree of evolution, there is an impact on patients' quality of life regarding functional dependence, social and family relationships, general assimilations of the environment, their physical and psychological condition, and among others. It is a disease of progressive and fatal character, with causes still little known [2]. The initial symptoms of ALS are muscular weakness, myo fasciculations, dysarthria, dyspnea, atony, dysphagia, amyotrophy, and respiratory symptoms in about 5% of cases [3,4]. In the terminal phase, most of the cases of death related to ALS arise from complications originating from respiratory failure, a condition that characterizes the final phase of the disease [5].
Diagnosis begins with physical examination, complementary and specific exams for confirmation [6]. As established by the World Federation of Neurology, the diagnosis should be made through clinical examination, electromyography, assessment of neurogenic abnormalities and progression of the clinical picture, and differential diagnosis [3]. According to the diagnosis and degree of degeneration of motor neurons, the diagnosis can be subclassified as: Definitive ALS, probable ALS, probable ALS with laboratory support, possible ALS, and ALS suspected [1,3,7,8].
Studies have demonstrated advances in understanding the development of ALS by sequencing the genes involved as superoxide dismutase 1 (SOD1) and vesicle-associated protein B (VAPB) [4]. The current treatment used as a palliative to ease the symptoms and difficulties arising from the disease and increase the survival time. In this context, the most effective drug in use is riluzole, which discretely postpones the degeneration of motor neurons. However, experimentally, there are gene therapies with gene insertion techniques capable of reducing the mutant SOD1 people and thereby decrease the degeneration of motor neurons, bringing various benefits to patients [9-13].
ALS is a highly debilitating disease, since the degeneration of motor neurons, besides the paraplegia characterizing the disease, causes serious symptoms, mainly respiratory, that end up reducing the survival and quality of life of the affected individuals. However, a definitive or highly effective treatment has not yet been developed. Recent research with gene therapies, still experimental, with techniques of insertion of genes, seek to benefit such individuals. Thus, the present theme is of great importance for the evaluation of the genetic profile associated with ALS, both for diagnosis and for the investigation of the genes involved in the pathological process that triggers the disease. Understanding the cause of this process can pave the way for more promising treatments. This bibliographic research aimed to describe the molecular processes, such as the main gene mutations and altered proteins, associated with the development of ALS.
METHODOLOGY
The present work is a bibliographical review, with a narrative, exploratory and descriptive character, in which information available in scientific databases is presented and discussed, aiming to analyze and evaluate articles published in scientific journals related to ALS. A retrospective study was carried out, through a bibliographical review for the search of publications and scientific articles on the subject, published in the period from 2012 to 2017, in the national and international literature. Content-based analyzes were performed by consultation at the Virtual Health Library, Google Academic, PubMed, and Scientific Electronic Library Online.
Information collection took place from March to August 2017. As a criterion for inclusion, published papers on the subject were considered, preferably from 2012 to 2017, which addressed subjects pertinent to the research. A total of 3360 articles were found, of which 39 were selected, according to their relevance to the composition of the present study. A thorough reading of the articles found in the bibliographic survey was done, according to the subjects. After analysis of the sources of information above, the articles that presented the terms ALS and the possible pathological mechanisms were selected. Articles older than 5 years were excluded from the selection.
LITERATURE REVIEW
ALS, considered one of the most serious neurodegenerative diseases, has as its main characteristic the death of the lower motor neurons, which include neurons of the anterior horn of the spinal cord and its equivalents in the brainstem, responsible for the enervation of the brain bulb; and upper motor neurons originating from the motor cortex and branching to establish synaptic contact with the lower motor neurons [14]. The term ALS describes the stiffening (hence the word sclerosis) of the lateral (lateral) spinal cord, due to degeneration of the motor neurons, accompanied by muscular atrophy (amyotrophic) [1]. In the initial phase of the disease, there may be only one of the neuronal types mentioned, but with their progression both are degenerate. In axons, accumulations of neuronal filaments and proteins are observed, and the neuroglia presents in a proliferative state, resulting in the innervation of the muscles, which perish in atrophy [9].
ETIOPATHOGENESIS
The etiology of ALS has not yet been clarified in the literature, but there are several endogenous and exogenous risk factors suspected of contributing significantly to the development of the disease, such as smoking, large-scale physical activity, electrocution damage, exposure to heavy metals, toxins, pesticides, insecticides, and other chemicals. There is also the race factor involved since the disease has a higher incidence in Caucasian individuals [15]. However, it must be considered that such risk factors are not determinant for the development of ALS, may act concomitantly or associated with the genetic profile of the patient [16].
Regarding the genetic factors involved, studies in mice indicated that SOD1 transgenes may be involved with the gene changes that lead to ALS. The enzyme SOD1 is responsible for the elimination of the superoxide anion, acting as a form of defense against the toxicity of glutamate, a toxic neurotransmitter [9]. In ALS, as the enzyme SOD1 is changed, becoming an unstable molecule and prone to aggregation, elimination of the glutamate ceases to exist, and it is free to participate in the destruction of motor neurons. This aberrant alteration also provides a reduction of axonal synapse, mitochondrial disorders, and poor ATP production. Mutations in other genes may also be involved in the processes associated with ALS, such as the tar binding DNA protein (TARDBP) encoding tar binding DNA protein 43 KDa (TDP43), fused in sarcoma (FUS), and VAPB [9,17].
HISTOPATHOLOGY
This disease is due to degeneration of the upper and lower motor neurons, which are replaced by microglial activation and astroglia proliferation. In axons are accumulations of neuronal filaments and aggregates of proteins, called spheroids. Other microscopic findings are: Lipofuscin, lipid pigments, Bunina corpuscles, spherical and eosinophilic structures, and inclusions of ubiquitin[9,15]. The pyramidal cells of the motor cortex, the myelinated fibers of the anterior and lateral spinal cord, the brainstem, and the cerebellum are degenerate, as well as the hypoglossal nucleus. Onuf's nucleus, ocular and pathetic motor nerves usually remain unharmed, being reached only in the pre-terminal phase of ALS. The neuronal destruction causes a lack of muscle fibers, which cause the appearance of fascicular formations that cause the absence of muscular activity, causing muscle atrophy [15,18]. Already pleocytosis is present, with a predominance of neutrophils and IgM viral antibodies are found [11].
CLINICAL MANIFESTATIONS
In general, the initial ALS symptoms correspond to muscle weakness, myo fasciculations, dysarthria, dyspnea, atony, dysphagia, amyotrophy, and in about 5% of the cases respiratory symptoms [3,4]. The clinical manifestations of ALS are related to the region of onset of the disease: Bulbar, cervical, or lumbar [7].
Individuals with bulbar onset ALS, that is, with degeneration of the lower motor neurons, which generates bulbar or higher paralysis, which generates pseudobulbar paralysis, may present dysarthria, dysphagia, dyspnea, facial paralysis, difficulty in palatal, and tongue movement, which is atrophied, emotional instability, and reflexive mandibular enlargement [5,15]. On the other hand, individuals with cervical onset ALS, with degeneration of the lower and upper motor neurons responsible for the movement of the trunk and upper limbs, may present amyotrophy and muscular weakness in the upper limbs with myo fasciculations and the reflexes may be intensified or reduced [1,6]. Individuals with lumbar onset ALS, with degeneration of lower and upper motor neurons responsible for lower limb movement, may present weakness in the lower limbs, lowered foot, and cramps [1,7].
DIAGNOSIS
The diagnosis should be made according to the World Federation of Neurology through El Escorial Revisited (1998), following the established criteria [1,3,8] Signs of ALS are: Degeneration of lower motor neurons (LMN) through electromyography, clinical, and neuropathological examination; degeneration of upper motor neurons (UMN) by clinical examination; and progression of ALS, observed by the spread of symptoms from one region to another. [1,3,8]. Furthermore, the absence of pathological and/ or electrophysiological sign that another disorder may be the cause of motor neuron degeneration is associated with ALS; a neuroimaging signal that another disorder may explain the clinical and electrophysiological findings; involvement of the sensorial and autonomous system [1,3,8]. The diagnosis is supported by fasciculation in a region; there are neurogenic mutations in the electroneuromyography, with normal conduction velocity, and without restriction [3].
Given the above criteria, the diagnosis can be subclassified as below:
- Definitive ASL: Clinical signs of degeneration in NMI and NMS in three regions, one bulbar and two cervical;[1,3,7,8].
- Probable ALS: Clinical signs of NMI and NMS degeneration in two regions, with the presence of some bulbar signal;[1,3,7,8]
- ASL likely with laboratory support: Signs of NMI and NMS degeneration in one region, supported with electromyography of the nervous system in two muscle segments;[1,3,7,8]
- Possible ASL: Signs of NMI and NMS degeneration in one region, or NMS in two, or three regions;[1,3,7,8]
- ASL suspected: Sign of NMI or NMS degeneration in a region [7].
Despite the existing criteria for the diagnosis of ALS, there is still a delay and risk of errors in the diagnosis, with an average time of 14 months to confirm this from the first consultation. Studies have shown about 10% false positive results at diagnosis in the past decade, due to the fact that clinical manifestations at the onset of ALS may be confounded with other diseases such as post-polio syndrome, monoclonal gammopathies, endocrinopathies, adrenoleukodystrophy, primary lateral sclerosis, benign fasciculation, lymphoma, myelopathies, HIV1 infections, HTLV1, varicella zoster, brucellosis, borreliosis, syphilis and lymphoreticulosis, and poisoning by metals such as lead, mercury, and aluminum 1 [5-19].
CURRENT TREATMENT, PROGNOSIS, AND COMPLICATIONS
The prognosis of ALS is often fatal, and there is currently no effective treatment to stop the deterioration of motor neurons. Only palliative care is indicated to improve the quality of life of the patient, alleviating the symptoms and difficulties arising from the disease. The main palliative care is those related to nutritional support (enteral feeding and gastronomy) and respiratory (non-invasive home ventilation, invasive ventilation, and tracheostomy), and in the terminal phase of the disease, and major Medicare care becomes necessary [1,10,12].
Riluzole (100 mg/day) is the only drug approved by the Food and Drug Administration to be administered in patients with ALS, with relative efficacy proven to discreetly increase patients'survival by 2-4 months, as it todelaythedeterioration of motor neurons, but without altering the course of the disease. The cause of postponement of neuronal deterioration from riluzole use is poorly understood, but it is suspected to be caused by reduced release of glutamate. However, the side effects of riluzole include nausea, dizziness, increased liver enzymes, and weight loss [9-12].
Other palliative treatments include the use of muscarinic anticholinergic medications in cases of sialorrhea complications, physiotherapy in conjunction with the use of crutches and walkers to improve mobility and avoid contractures, use of orthoses for fallen feet and splints for hand extension and use of coughing devices to clear the airways [9,11].
Other drugs such as celecoxib, coenzyme Q-10, creatine, gabapentin (marketed in Brazil as neurontin), cialis neurotrophic factor, brain-derived neurotrophic factor, insulin-like growth factor, lamotrigine, lithium, minocycline, valproic acid, verapamil, and xaliproden were shown to be ineffective in ALS. However, studies involving glial-derived neurotrophic factor had encouraging preclinical results, and experiments with Vitamin E brought a small clinical improvement to treated patients [3,11].
In the terminal phase, for the treatment of dyspnea and pain, opioids are used together with benzodiazepines, in cases of excessive anxiety. Already patients with insomnia and confusion caused by hypercapnia use neuroleptics such as chlorpromazine. In cases of proven hypoxia, the use of oxygen therapy is recommended [6].
The complications of ALS are flaccid paresis, reduction and later absence of reflexes, spastic paresis, reduction of nerve conduction velocity, which will result in respiratory and motor insufficiency, progressive bulbar paralysis, speech problems, sialorrhea, venous thrombosis, cramps, depression, anxiety, paraplegia, hypercapnia, alveolar hypoventilation among others [20]. Respiratory failure and its complications are responsible for about 85% of cases of ALS deaths [5].
MOLECULAR BASES INVOLVED IN THE DEVELOPMENT OF ALS
Most cases of ALS are sporadic, with no hereditary association. About 90-95% of cases occur in patients over 50 years of age. In cases of familial ALS, other members are affected in the family, with first- or second-generation kinship. In the family ALS, early onset of the disease occurs, with an initial phase from the lower limbs and life expectancy related to the involved genotypes [21] It has high heritability, attached to the X chromosome and may be autosomal recessive in juvenile cases, and autosomal dominant, in cases beginning in adulthood. The most common gene mutations involved in familial ALS are the genes SOD1, FUS, TARDBP, and chromosome 9 open reading frame 72 (C9orf72) [Table 1]. Almost half of the patients with ALS with mutations in the SOD1 and FUS genes present clinical manifestations from the age of 50 years [22,23] Genetic subtypes and age at onset of clinical manifestations are well correlated. Concerning bulbar symptoms, familial ALS is more exacerbated than in sporadic ALS [22-24].
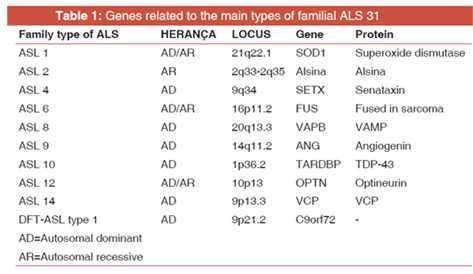
About 22 locus associated with mutations (ASL 1 - ASL 16) were identified for the family ALS, with some being:
SOD1 (ASL 1), C9orf72 (ASL1), ALS2 (alsin - ASL 2),SETX (senataxin - ASL 4), SPG11 (spatacsin 11 - ASL 5), FUS (ASL 6), VAPB (ASL 8), ANG (angiogenin - ASL 9), TARDBP (ASL 10), FIG4 (ASL 11), OPTN (optineurin - ASL 12), ATXN2 (ataxin-2-ASL 13), valosin-containing protein, and ubiquilin2-ASL 15 (UBQLN2). For ALS 3 and ASL 7, mutations occur on chromosomes 18q21 (ALS 3) and 20p13 (ALS 7) [25]. In sporadic ALS, mutations were also identified in the TARDBP, C9orf72, SOD1, ANG, FUS, OPTN, and SETX genes. In this article, only the main ones of these mutations will be discussed [24,26].
Only in about 5-10% of all cases of ALS were the genes responsible for the disease identified. 40% of the cases are caused by mutations in the C9orf72 gene, 20% in the SOD1 gene, 4-5% in the FUS and TARDBP genes, and the remainder is caused by mutations in other genes such as ALS2, SETX, SPG11, VAPB, ANG, FIG4, OPTN, ATXN2, and UBQLN2.Only the genes of higher frequency and clinical importance (C9orf72, SOD1, FUS, TARDBP, and VAPB) were selected for this study, and these will be presented below according to the type of ALS they cause [21,24].
Despite the identification of the 22 locus, there are still 32% of cases of familial ALS and 11% of cases of sporadic ALS with a genetic diagnosis not yet defined [25]. In this context, there are still several points to be clarified regarding the role of the mentioned genes, since some of them do not have a single process that causes the disease, but disease-modifying mechanisms [27].
The SOD1 gene, encoding the antioxidant enzyme Cu+2/ Zn+2 SOD, was the first to be identified in cases of ALS and is the second largest form of familial ALS, accounting for up to 20% of ALS cases and up to 7% of ALS cases. Patients with a mutation in this gene have ALS with a predominant onset in the lower limbs and may also present bulbar onset in some cases [21,24,28] SOD1 is located on chromosome 21 (locus 21q22.1), is 11kb long in DNA and consists of 5 exons, which encode the 153-amino acid enzyme, and is interrupted by 4 introns. To date, 150 ALS-causing mutations have been identified in these exons [24,28].
The analysis of phenotypic changes by mutations in SOD1 reveals that such phenotype depends on environmental and genetic factors. As an example, the SOD1D90A mutation that is recessive in Scandinavian patients can be cited but is dominant in other genetic groups. Patients with the SOD1A4V mutation have shorter survival, about a year and a half after diagnosis. This has a penetrance of 91%. The SOD1A89V mutation has incomplete penetrance and affects variable age ranges. The SOD1|113T mutation has diversification in all its aspects, including age, progression, clinical manifestations, and penetrance [24].
The enzyme encoded by this gene is present in the cytoplasm, nucleus, lysosomes, and between mitochondrial membranes. It has the function of capturing copper and zinc ions to form a homodimer, capable of dismutation of superoxide, a toxic radical, in oxygen, and hydrogen peroxide, which are metabolized in water and oxygen by the enzymes glutathione peroxidase and catalase [Figure 1] [29].

Due to the mutations occurred, the enzyme SOD1 has its reduced activity, leading to insufficient dismutation of the superoxide radical by the altered enzyme. Consequently, the accumulation of these radical results in oxidative stress toxic to motor neurons. The enzyme is prone to incorrect folding due to the mutation and aggregate form in the cellular cytoplasm of the neurons, resulting in the sequestration of essential proteins to neural cell metabolism, interruption of the ubiquitin/proteasome system and axonal transport, in the mitochondria rupture, of the cytoskeleton. All these factors culminate in neural death [28].
The exact mechanism by which SOD1 mutations trigger ALS is still unknown, but there are several hypotheses as to how they contribute to motor neuron death, including protein aggregation due to mutant SOD1 folding, oxidative stress, mitochondrial and endoplasmic reticulum dysfunction, glutamate excitotoxicity, microglial inflammation and activation, and abnormal axonal transport [24].
Mutations in SOD1 have variability regarding the age and place of onset of clinical manifestations and penetrance. The mutation in the most common SOD1 gene is D90A, which does not cause functional loss of the protein. It remains stable under denaturing conditions. In this case, the loss of lower limb motility may take years to emerge [28].
ASL 6 is caused by mutations in the FUS gene, located on chromosome 16 and with 16p12.1-q21 mutation locus, and accounts for up to 4-5% of cases of familial ALS. To date, more than 50 autosomal recessive or dominant mutations have been identified that cause familial ALS for this gene. Most of the mutations are missense, and the others are frameshift or nonsense, there is a correlation with the involvement of the TDP-43 protein[21,28]. The predominance of symptoms is associated with lower motor neurons, with tremors. There is no impairment of the bulbar region or cognitive impairment. The disease starts early in cases where neural cytoplasm presents basophilic and compact [21,24].
The FUS gene is 9 kb long, with 15 exons, and is responsible for encoding the same-name protein that binds DNA/ RNA to the cell nucleus. The FUS nucleoprotein has as functions the regulation of the transcription; repair the DNA, splicing, transport between the nucleus and cytoplasm, and development of the mRNA. In the central nervous system, it regulates the transport of mRNA to dendrites and the activation of glutamate receptors [28,30]. As markers of oxidative stress, FUS and TDP-43 inclusions were found in the spinal cord of patients with ALS, indicating a relation of these inclusions with the oxidative stress characteristic of ALS, both familiar and sporadic. Mutations in the FUS gene cause the mentioned cytoplasmic inclusions and loss of the physiological functions of this nucleoprotein [28].
VAMP Associated Protein A (VAPs), encoded by the gene of the same name found on chromosome 18, VAMP and VAPC, are encoded by the gene. The VAPP (Associated Membrane- Associated Proteins) proteins are found in the membrane of the endoplasmic reticulum. VAPB [4,21].
The VAPB gene, located on chromosome 20 (locus 20q13.3), 57.7 kb in length and composed of 6 exons, is responsible for encoding vesicle-associated membrane protein-associated protein B (VAMP) functions of intracellular vesicle transport, lipid transport, and reaction in the protein [4,28]. VAMP protein has three specific domains: N-terminal globular domain that shares its sequence with the protein major sperm protein, being called the domain MSP; coiled-coil domain and C-terminal transmembrane domain which grants the homo or heterodimerization of proteins of this type [4].
The mutation that promotes the exchange of proline 56 by the serine from the MSP domain (P56S) denatures the protein, disrupting its three-dimensional structure and making it prone to aggregation. This change generates instability in the interaction of VAMP with other endoplasmic reticulum proteins, including tubulin and GAPDH and is reported as responsible for ALS 8. The P56S mutation in VAPB generates a mutant protein that compromises the intracellular membrane transport and secretion system of motor neuron, causing loss of neural signals, and consequently cell death [4,28].
The dominant character of ALS 8 is due to the P56S mutation generates a dominant negative effect. VAPB is present in all cells of the human organism, but this mutation affects only motor neurons, probably because the other cell types do not require the same amount of VAPB to maintain their survival, or VAPB has a specific function still unknown in the motor neurons [4,28].
The mutant VAMP protein forms fixed aggregates in the endoplasmic reticulum, which promotes the reduction of this organelle, the difficulty in the transport of lipids that are retained and the sequestration of functional proteins in inclusion bodies, which culminate in motor neuron degeneration. In addition to impairment of function, the mutant VAPB becomes toxic, since its inclusions may cause changes in the proteasomes and alter the means of protein degradation, coupled with a pathogenic mechanism due to the toxicity of mutant protein aggregates and inhibition of transport mitochondrial, which affects the regulation of kinesin [4,28].
ASL 10 accounts for up to 4-5% of cases of familial ALS and are a rare autosomal familial form of ALS, early onset related to missense mutations in the TARDBP gene, responsible for encoding the TDP-43 protein (TAR DNA-binding protein 43), which is a protein ligand of DNA and RNA, a modulator of transcriptional regulation. This type of ALS represents about 5% of cases of familial ALS and 2% of sporadic cases. Mutations in TARDBP have already been clinically established, with rapidly progressing G298S mutations and A315T and M337V mutations with longer survival [21,23,31].
TDP-43 is a heterogeneous ribonucleoprotein protein. It is present in several biological processes, including gene transcription, regulation of splicing, transport, and stabilization of mRNA molecules. It is found in cytoplasmic inclusions of ubiquitin in patients with ALS, being hyperphosphorylated and cleaved to generate abnormal C-terminal fragments. In normal motor neurons, TDP-43 is present in the nuclei, but in the development of ALS, it becomes absent in nuclei of neurons with cytoplasmic inclusions of ubiquitin [28,30].
The pathogenic mechanisms involving the TDP-43 protein include toxicity by aggregation of this protein or gain of toxic function due to mutations in the TARDBP 31 gene. Many mutations have been described and have already been well established, and all are found in the C-terminus, encoded by exon 6. The Q331K mutation generates hyperphosphorylation of the TDP-43 protein due to the creation of another protein kinase A. The G294A mutation is involved in the binding of RNA binding and gene suppression. The A382T mutation is related to the predominance of ALS of the lower motor neurons, with onset in the distal muscles of the limbs, and with disease progression, spreads to the proximal muscles [28].
Mitochondrial dysfunction is also present in ALS 10, confirmed in yeast and mammalian cell models with mutations in the TARDBP gene, which presented an abnormal mitochondrial structure [28,30].
Frontotemporal dementia - ALS type 1 is the most common type of familial ALS, accounting for up to 40% of cases of familial ALS. It has an autosomal dominant character and is derived from the repetition expansion of the hexanucleotide (GGGGCC) of the C9orf72 gene (locus 9p21.2), pertinent to chromosome 9 [32].
Haploinsufficient causes loss of function and toxic mechanisms are present. The mutation in the C9orf72 gene generates abnormal neuronal membrane, endosomal traffic derived from proteins such as ubiquilin-2, HNRNPA1 (heterogeneous nuclear ribonucleoprotein A1) and HNRNPA2B1 (heterogeneous nuclear ribonucleoprotein A2/ B1), mutant RNA with gain toxic function, and abnormalities in the ubiquitin-proteasome. Axonal transport becomes deficient and abnormal hexanucleotide causes the formation of hybrid DNA/RNA with connections of aborted transcripts, generating nucleolar stress, and neuronal apoptosis. It is also observed the absence of the ATG start codon in the expression transcripts, which causes the formation of repeated proteins of dipeptides, toxic to the neuronal DNA [32,33].
Spinal cord motor neurons and glia cells present with cytoplasmicinclusionsofubiquitinandubiquilin-2 [22]. Patients with DFT-ALS have Lewy body dementia, progressive supranuclear palsy, corticobasal degeneration, movement disorders, and psychiatric disorders [21].
FINAL CONSIDERATIONS
ALS is a progressive and fatal neurodegenerative disease that affects the quality of life of patients, who need family, medical, and psychological support to face the limitations that this disease provides throughout their progress. Until today, a promising treatment that stops the disease progression and reverses the damage caused to motor neurons has not been officially developed, since all the existing treatments are only intended as palliative care to improve the patient's quality of life and increase his survival. Regarding promising treatment, there is only experimental research, such as gene therapy.
In recent years, with the discovery of several genes associated with the development of ALS, the mechanisms involved in this pathological process began to be clarified, contributing to the search for more promising treatments and faster diagnosis. In this work, the main genes (SOD1, FUS, VAPB, TARDBP, and C9ORF72) were approached with greater frequency and clinical importance and the pathological mechanisms triggered by each one was presented, culminating in irreversible lesions to motor neurons, often derived from stress oxidative stress due to the formation and aggregation of mutant proteins, and in progressive muscular atrophy. It is extremely important that research on ALS continues so that all the mentioned mechanisms are unveiled and that a definitive treatment is finally found.
REFERENCES
- Lavoisier LN, Carolina CA. Dysarthria and quality of life in patients with amyotrophic lateral sclerosis. Rev CEFAC 2017;19:664-73. Available from:
http://www. scielo.br/scielo.php?script=sci_arttext&pid=S1516- 18462017000500664&lng=en. [Last cited on 2018 Oct 03]. - Silva NP, Martins LJ, Ferreira TB, Cavalcanti FA. Correlation between functional independence and quality of life of patients with amyotrophic lateral sclerosis. Cad Ter Ocup UFS Car 2014;22:507-13.
- Quadros AA, Oliveira AS, Silva HC, Chieia MA, Pereira RD, Fernandes E, et al. Abrela-Associacao Brasileira de Esclerose Lateral Amiotrofica; 2013. Available from: http://www. abrela.org.br/default.php?p=principal.php. [Last citado on 2016 Sep 23].
- Alan ER, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014;17:17.
- Ahmed RM, Newcombe RE, Piper AJ, Lewis SJ, Yee BJ, Kiernan MC, et al. Sleep disorders and respiratory function in amyotrophic lateral sclerosis. Sleep Med Rev 2016;26:33-42.
- Vilabril F, Costa JV, Santo VE, Miranda C, Pires E, Dias L. Impact of a rehabilitation program in the overall survival of amyotrophic lateral sclerosis patients. Ann Phys Rehabil Med 2018;61:e254.
- Brasil MS. Protocolo Clinico e Diretrizes Terapeuticas- Esclerose Lateral Amiotrofica; 2015.
- Madureira CD. Diagnostico Diferencial de Esclerose Lateral Amiotrofica: A Proposito de um Caso Clinico (Doctoral Dissertation, Universidade da Beira Interior); 2012. Available from:
http://www.ubibliorum.ubi.pt/handle/10400.6/1098. - Hauser S, Josephson S. Neurologia Clinica de Harrison-3. AMGH Editora; 2015. Available from: https://www. books.google.com.br/books?hl=pt-BR&lr=&id=4d ObBQAAQBAJ&oi=fnd&pg=PR1&dq=Brown+RH+Jr
- Moura MC. Aspectos Epidemiologicos, Prognosticos E Tratamento Da Esclerose Lateral Amiotrofica.Universidade de Brasilia; 2016. Available from: http://www.repositorio.unb.br/ handle/10482/19930.
- Greenberg DA, Aminoff MJ, Simon RP. Neurologia clinica. 8th ed. Porto Alegre: AMGH; 2014.
- Schlindwein-Zanini R, Queiroz LP, Claudino LS. Aspectos Neuropsicologicos Da Esclerose Lateral Amiotrofica: Relato de caso. Arq Catarinenses Med 2016;44:62-70.
- Neto MM. Terapia Genica e ELA. 2015.
- Handy CR, Krudy C, Boulis N, Federici T. Pain in amyotrophic lateral sclerosis: A neglected aspect of disease. Neurol Res Int; 2011. Available from: https://www.hindawi.com/journals/ nri/2011/403808/abs.
- Godinho VC. Esclerose Lateral Amiotrofica: Revisao Bibliografica da Patofisiologia (Doctoral Dissertation, Universidade da Beira Interior); 2013. Available from: http:// www.ubibliorum.ubi.pt/handle/10400.6/1402.
- Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, et al. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 2015;6:171.
- Nascimento CF. Caracterizacao das alteracoes da proteina TDP-43 durante o envelhcimento normal: Uma analise em cErebros humanos postmortem. Brazil: Universidade de Sao Paulo; 2015. p. 143.
- Daroff RB, Jankovic J, Mazziotta JC, Pomeroy SL, Bradley WG. Bradley's neurology in clinical practice. Philadelphia, PA: Saunders; 2012.
- Galan Davila, L. Diagnostico de ELA. Madrid: Hospital Clinico San Carlos. 2015.
- Orsini M, Oliveira AB, Nascimento OJ, Reis CH, Leite MA, de Souza JA, et al. Amyotrophic lateral sclerosis: New perpectives and update. Neurol Int 2015;7:5885.
- Souza PV, Pinto WB, Chieia MA, Oliveira AS. Clinical and genetic basis of familial amyotrophic lateral sclerosis. Arq Neuropsiquiatr 2015;73:1026-37.
- Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH, et al. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol 2012;124:339-52.
- Leblond CS, Kaneb HM, Dion PA, Rouleau GA. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp Neurol 2014;262 Pt B:91-101.
- Chen S, Sayana P, Zhang X, Le W. Genetics of amyotrophic lateral sclerosis: An update. Mol Neurodegener 2013;8:28.
- Vucic S, Rothstein JD, Kiernan MC. Advances in treating amyotrophic lateral sclerosis: Insights from pathophysiological studies. Trends Neurosci 2014;37:433-42.
- Prado LG. Perfil Clinico De Pacientes Portadores De Esclerose Lateral Amiotrofica Acompanhados Em Centros De Referencia De Belo Horizonte. MG, Brasil 2015. Available from:
http://www.bibliotecadigital.ufmg.br/dspace/handle/1843/ BUBD-9VZR3Z?show=full - Su XW, Broach JR, Connor JR, Gerhard GS, Simmons Z. Genetic heterogeneity of amyotrophic lateral sclerosis: Implications for clinical practice and research. Muscle Nerve 2014;49:786-803.
- Mancuso R, Navarro X. Amyotrophic lateral sclerosis: Current perspectives from basic research to the clinic. Prog Neurobiol 2015;133:1-26. Available from:
https://www.sciencedirect. com/science/article/pii/S0301008215000787. - Saccon RA, Bunton-Stasyshyn RK, Fisher EM, Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013;136:2342-58.
- Finelli MJ, Liu KX, Wu Y, Oliver PL, Davies KE. Oxr1 improves pathogenic cellular features of ALS-associated FUS and TDP-43 mutations. Hum Mol Genet 2015;24:3529-44.
- Corcia P, Valdmanis P, Millecamps S, Lionnet C, Blasco H, Mouzat K, et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology 2012;78:1519-26.
- Souza PV, Pinto WB, Oliveira AS. C9orf72-related disorders: Expanding the clinical and genetic spectrum of neurodegenerative diseases. Arq Neuropsiquiatr 2015;73:246-56.
- Woollacott IO, Mead S. The C9ORF72 expansion mutation: Gene structure, phenotypic and diagnostic issues. Acta Neuropathol 2014;127:319-32.