Bone Marrow Histology is a Pathognomonic Clue to Each of the JAK2V617F, MPL,515 and Calreticulin Mutated Thrombocythemia in Myeloproliferative Neoplasms
Hendrik De Raeve1,2, Karel Fostier3, Francisca Valster4, Vincent Potters5, Yango Kim5,6, Myungshin Kim5,6, Zwi Berneman7, Wilfried Schroyens7, Jan Jacques Michiels7,8
2.Department of Pathology, Belgium and University Hospital Brussels, Laarbeeklaan 101, B-1090, Brussels, Belgium.
3.Department of Hematology, University Hospital Brussels, Laarbeeklaan 101, B-1090, Brussels, Belgium.
4.Department of Internal Medicine, Bravis Hospital, Boerhaaveplein 1, 4624 VT, Bergen op Zoom, Netherlands.
5.Department of Pathology, Bravis Hospital, Boerhaveplein 1, 4624 VT, Bergen op Zoom, Netherlands.
6.Department of Pathology, Bloodcoagulation and Vascular Medicine Specialist, Goodheart Institute in Nature Medicine and Health, Erasmus Tower, Veenmos 13, 3069 AT Rotterdam, Netherlands.
7.Department of Hematology, University Hospital Antwerp, Wilrijkstraat 10, B-2650, Edegem, Belgium.
8.Dutch MPN Foundation and International Collaboration and Academic Research on Myeloproliferative Neoplasms, Goodheart Institute, Erasmus Tower, Veenmos 13, 3069 AT, Rotterdam, The Netherlands.
Citation: De Raeve H, Fostier K, Valster F, Potters V, Kim Y, Kim M, Berneman Z, Schroyens W, Michiels JJ. Bone Marrow Histology is a Pathognomonic Clue to Each of the JAK2V617F, MPL,515 and Calreticulin Mutated Thrombocythemias in Myeloproliferative Neoplasms. Clin Res Hematol 2018;1(2):1-7.
According to the World Health Organization and Clinical Laboratory Molecular and Pathological criteria bone marrow pathology in JAK2V617F mutated trilinear myeloproliferative neoplasm (MPN) patients essential thrombocythemia (ET) and polycythemia vera are indistinguishably featured by clustered medium to large pleomorphic megakaryocytes and increased cellularity (60-90%) due to increased erythropoiesis and megakaryopoiesis. MPL,515 mutated ET is the second distinct clonal MPN characterized by thrombocythemia in a normocellular bone marrow showing clustered increased large to giant mature megakaryocytes with staghorn-like hyperlobulated nuclei. Calreticulin (CALR) mutated hypercellular thrombocythemia associated with prefibrotic megakaryocytic, granulocytic myeloproliferation (MGM) recently became the third distinct MPN featured by dense clusters of immature megakaryocytes with cloud-like nuclei. Bone marrow pathology in newly diagnosed MPN patients appears to be a pathognomonic clue for diagnostic differentiation between JAK2V617F mutated trilinear MPN, MPL,515 normocellular thrombocythemia, and CALR thrombocythemia with MGM characteristics followed by secondary reticulin fibrosis. Their natural histories clearly differ featured by an increase of erythro/granulopoiesis and cellularity in JAK2V617F, decrease of erythropoiesis and cellularity in MPL,515 and increase of dual megakaryo/granulopoiesis and cellularity in CALR mutated MPN.
Bone marrow pathology, calreticulin mutation, essential thrombocythemia, JAK2 mutation, MPL mutation, myelofibrosis, polycythemia vera, reticulin fibrosis
INTRODUCTION
The clinical and pathological features for prodromal erythrocythemia and classical polycythemic stages of polycythemia vera (PV) are variable and featured by increased erythrocytes above 6X1012/L, increased leukocyte alkaline phosphatase (LAP) score (increased CD11b expression), normal or increased platelets, leukocytes, and spleen size, and by characteristic bone marrow features with increased pleomorphic large megakaryocytes and erythropoiesis [Table 1] [1,2]. The peripheral blood findings in "true" essential thrombocythemia (ET) without features of PV are featured by high platelet counts, normal values for hemoglobin, hematocrit, erythrocyte and white blood cells, and normal values for LAP score, LDH and no or minor splenomegaly despite platelet counts above 1000X109/L at time of first presentation [3-5]. The megakaryocytes in "true" ET are larger than in ET preceding PV and classical PV [Table 1] [3-6]. The Hannover Bone Marrow Classification of the MPDs distinguished three primary prefibrotic MPDs ET, PV, and primary megakaryocytic granulocytic myeloproliferative (PMGM) from the advanced fibrotic stages of MPD [7]. Bone marrow pathology in PV is typically featured by large pleomorphic megakaryocytes with hyperploid nuclei in a hypercellular bone marrow due to increased erythropoiesis or increased trilinear erythrocytic, megakaryocytic, granulocytic myeloproliferation (EMGM)[6] Michiels and Thiele defined normocellular "true" ET as a distinct myeloproliferative neoplasm (MPN) entity different from classical PV and hypercellular ET associated with prefibrotic primary myelofibrosis (pPMF), which is consistent with primary megakaryocytic granulocytic myeloproliferation [PMGM, Table 1] [5,6]. Georgii et al. labeled pPMF as the third MPD entity of chronic or primary MGM (CMGM/PMGM) in the absence of reticulin or collagen fibrosis in bone marrow biopsy material [7,8]. Michiels et al. replaced the term CMGM by primary MGM (PMGM) as the third JAK2 wild-type MPN without features of PV or CML [6]. Hypercellular JAK2 wild-type ET associated with PMGM is dominated by an increase of clustered atypical dysmorphic megakaryocytes due to increases of cellular and nuclear size and bulky nuclei with clumsy lobuli and irregular roundish shaped forms (so-called cloud-like nuclei), which are never described in JAK2V617F mutated ET and PV [Table 1] [4-6]. The natural history of JAK2V617F thrombocythemia and PV is best reflected by the increase of erythropoiesis, granulopoiesis, and cellularity followed by the degree of anemia, splenomegaly, bone marrow cellularity, and an increase of reticulin and collagen fibrosis [7,8].

BONE MARROW PATHOLOGY IN MPL,515 MUTATED NORMOCELLULAR THROMBOCYTHEMIA
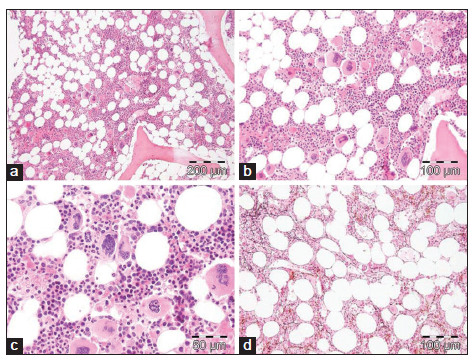
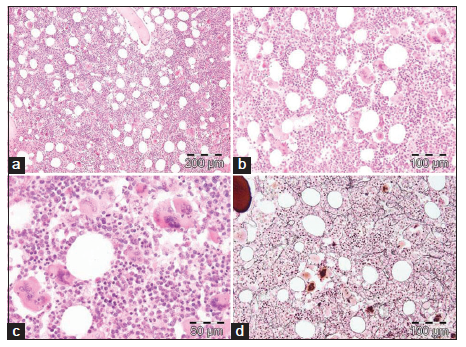
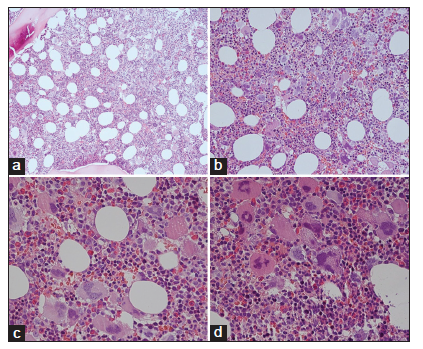
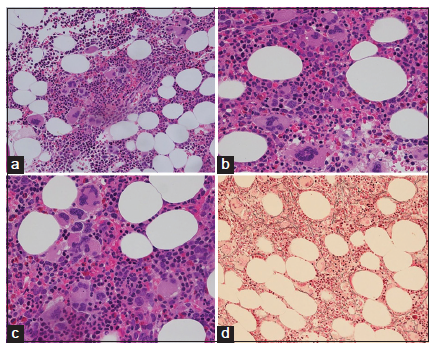
Bone marrow histology from a patient with thrombocythemia carrying the MPLW515L mutation displayed clusters of large megakaryocytes with a greater number of giant megakaryocytes with hyperlobulated stag-horn nuclei in normal cellular bone marrow and no increase of erythropoiesis [Figures 1-4] [9-13]. We here extend our previous descriptions on the differential diagnostic significance between patients with MPL,515 mutated (n = 12) versus JAK2V617F mutated trilinear MPN6. The presence of clustered small and giant megakaryocytes with deeply lobulated staghorn like hyperlobulated nuclei [Figures 1-4] in MPL,515 mutated thrombocythemia is not seen in JAK2V617F mutated ET and PV [Figures 5 and 6] and calreticulin (CALR) thrombocythemia. The increase of erythropoiesis is not seen in MPL,515 mutated ET [Figures 1-4] [6]. MPL,515 mutated ET have no clinical, laboratory and bone marrow features of prodromal PV at diagnosis [Table 1], do not evolve into PV during follow-up. JAK2V617F mutated ET [Figure 5] show local increase of erythropoiesis in areas of loose clustered pleiomorphic megakaryocytes in normocelluar JAK2V617F mutated ET [Figure 5], whereas bone marrow is hypercellular due to increased erythropoiesis and megakaryopoiesis (EM) [12,13]. JAK2V617F mutated ET, prodromal PV, and classical PV have increased score for LAP stain, low serum EPO and pleomorphic medium-sized to large mature megakaryocyte morphology [Figures 5 and 6]. In contrast, MPL,515 mutated normocellular ET has normal values for LAP score, serum EPO and ferritin levels [3,6,9-11]. As demonstrated in Figures 2-4, the natural history of MPL,515 normocellular ET is best reflected by decreased cellularity due to decreased erythropoiesis and increase of reticulin fibrosis (RF) from Grade 0 to Grades 1, 2, and 3.
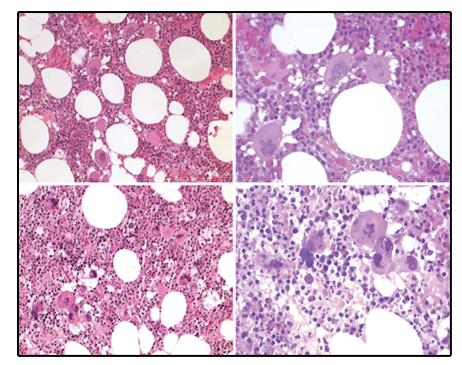
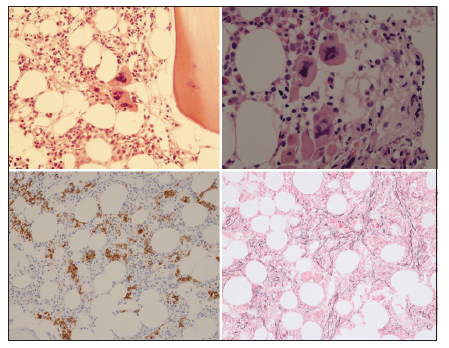
BONE MARROW HISTOLOGY IN CALR MUTATED ET AND MF
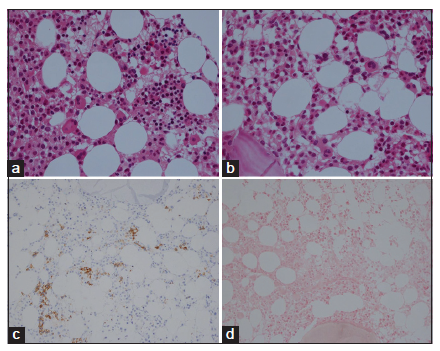
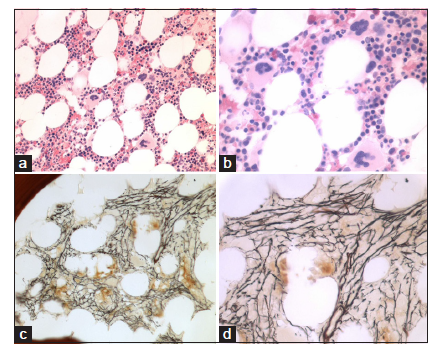
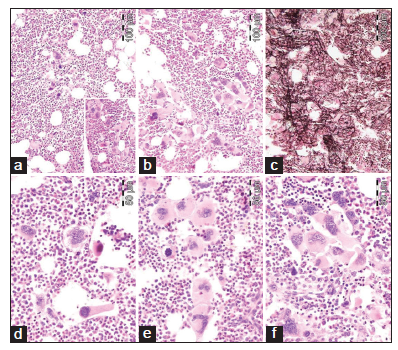
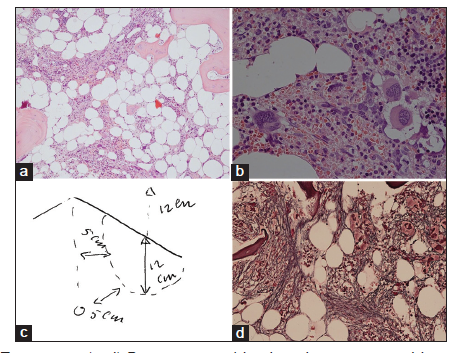
We found consistent bone marrow characteristics of hypercellular ET as the presenting feature of prefibrotic and early fibrotic or even advanced stages of PMGM in 13 consecutive newly diagnosed CALR positive ET cases (manuscript in preparation). 13 CALR mutated PMGM patients do not present present with ET complicated by aspirin-sensitive microvascular disturbances of erythromelalgic, cerebral and ocular ischemic manifestations (Sticky Platelet Syndrome) as the specific presenting manifestations of JAK2V617F and MPL,515 mutated myeloproliferative thrombocythemia. Bone marrow histology in prefibrotic CALR Thrombocythemia in Figure 7 show dysmorphic megakaryocytes with definite abnormalities of maturation with bulky (bulbous) hyperchromatic nuclei and some disturbances of the nuclear-cytoplasmic ratio consistent with CALR mutated PMGM, which are not seen in MPL,515 mutated ET [Figures 1-4] and also not in JAK2V617F mutated ET [Figure 5], prodromal PV and classical PV [Figure 5]. A 37-year-old woman (asymptomatic except fatigue) presented in 2004 with JAK2/MPL wild-type hypercellular ET associated with PMGM featured by platelets 1205X109/L, Hb 12.5 g/dl, leukocytes 18 X 109/L, borderline LDH, spleen size 13 cm on echogram (normal value < 12 cm) and PMGM bone marrow features with RF Grade 1/2 diagnosed according to the WHO - European clinical, molecular, and pathological criteria [6] [Figure 8 and Table 1]. 10 years later, this case was diagnosed as end-stage CALR mutated MF and treated by allogenic bone marrow transplantation in 2014. The three PMGM cases in Figures 7-9 clearly demonstrate that the natural history of CALR mutated PMGM, as the third distinct MPN entity, is featured by progressive anemia related to myeloid metaplasia of the spleen with splenomegaly, reduction of bone marrow erythropoiesis and progressive increase of RF Grade 1 to Grade 4 [14-18]. The natural history of CALR thrombocythemia and MF is best reflected by the degree of anemia, splenomegaly, bone marrow cellularity, and an increase of RF [Figures 7-10].
DISCUSSION
WHO-defined JAK2V617F mutated ET patients have PV-like morphological bone marrow changes of medium-sized to large pleomorphic megakaryocytes similar to our findings in newly diagnosed JAK2V617F mutated ET, prodromal PV patients, and PV patients [6,12,13]. JAK2V617F positive ET and prodromal PV patients usually have low serum EPO, increased LAP score, and slight to moderate increased bone marrow cellularity due to increased erythropoiesis. Increase of bone marrow EMG is the hallmark of classical prefibrotic PV with no or minor splenomegaly, whereas serum LDH levels, CD34+ circulating cells and spleen size are more pronounced in advanced JAK2V617F mutated ET (masked PV) or post-PV MF at high JAK2V617F mutation allele burden up to 90-100% [6,13]. Clustered large and giant megakaryocytes with hyperlobulated "staghorn" nuclei are rare in JAK2V617F mutated MPN, but typically present in MPL,515 mutated ET patients with no features of PV in the bone marrow and normal values for serum EPO, ferritin levels and LAP score [9,10]. The prevalence of MPL,515 mutated ET or MF patients ranges from 5% to 10% of the JAK2 wild-type MPN population [9-11].
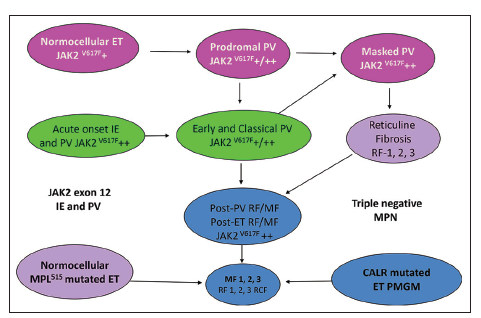
Dr. Kralovics and his team in Vienna Austria first discovered the occurrence of CALR mutation in 78 of 311 (25%) ET patients and in 72 of 203 (35%) MF patients and none of 382 PV patients [14]. Since 2013 CALR mutated thrombocythemia and MF became the third distinct MPN entity with no features of PV, which was rapidly confirmed by Nangalia et al [15] and by Rumi et al [16,17]. Nangalia et al. found somatic CALR mutations in 110 of 158 JAK2/MPL wild-type MPN, including 80 of 112 (70%) ET patients and 18 of 32 (56%) MF patients [15]. CALR mutations in the study of Nangalia et al. were identified in 10 of 120 (8%) MDS patients (RA in 5 of 53, RARS in 3 of 27, and RAEB-T in 2 of 27), and in one patient each with CMML and atypical CML [15]. The bone marrow pathological findings in the present study of CALR mutated thrombocythemia and MF in Figures 7-10 show dense clusters of large immature megakaryocytes with immature cloud-like nuclei, which clearly differ from the pleomorphic megakaryocytes in JAK2V617F mutated ET and PV, which indeed are clearly distinct from the giant megakaryocytes with hyperlobulated staghorn-like nuclei in MPL,515 mutated ET [Figures 1-4] [6]. Since 2014 Michiels et al. demonstrated that cases of hypercellular ET associated with PMGM belong to the third MPN of CALR-mutated thrombocythemia and MF without features of PV at time of diagnosis and during follow-up [6,14-20]. The updated 2018 WHO - Clinical Laboratory Molecular and Pathological (CLMP) criteria define five clonal mutually exclusive MPNs and transitional states at the laboratory and bone marrow level [Figure 11]: JAK2V617F mutated ET, prodromal PV, classical and advanced PV; JAK2 exon 12 mutated idiopathic erythrocytosis and PV; MPL,515 mutated thrombocythemia and MF; and CALR mutated thrombocythemia and MF and triple negative ET and MF [6,14-20]. The JAK2V617F positive trilinear MPDs, MPL,515 normocellular thrombocythemia, and CALR mutated thrombocythemia and MF mutually exclude each other [Table 1]. The natural history and MPN disease burden of each of the clonal MPNs is best reflected by the degree of anemia and splenomegaly on top of mutation allele burden, bone marrow cellularity and an increase of RF [6,15-20]. Prospective validation studies are warranted to confirm and amend the proposed WHO-CLMP classification.
REFERENCES
- Michiels JJ, Barbui T, Finazzi G, Fuchtman SM, Kutti J, Rain JD, et al. Diagnosis and treatment of polycythemia vera and possible future study designs of the PVSG. Leuk Lymphoma 2000;36:239-53.
- Michiels JJ, Kvasnicka HM, Thiele J. Doctor's Brochure 2004, Myeloproliferative Disorders Essential Thrombocythemia, Polycythemia Vera and Chronic Idiopathic Myelofibrosis. MPD. Available from: http://www.mpn-stichting.nl/doctors_ brochure_2004.pdf.
- Thiele J, Zankovich R, Schneider G, Kremer B, Fischer R, Diehl V, et al. Primary (essential) thrombocythemia versus polycythemia vera rubra. A histomorphometric analysis of bone marrow features in trephine biopsies. Anal Quant Cytol Histol 1988;10:375-82.
- Thiele J, Kvasnicka HM, Diehl V. Initial (latent) polycythemia Vera with thrombocytosis mimicking essential thrombocythemia. Acta Haematol 2005;113:213-9.
- Michiels JJ, Thiele J. Clinical and pathological criteria for the diagnosis of essential thrombocythemia, polycythemia vera, and idiopathic myelofibrosis (agnogenic myeloid metaplasia). Int J Hematol 2002;76:133-45.
- Michiels JJ, Berneman Z, Schroyens W, De Raeve H. Changing concepts of diagnostic criteria of myeloproliferative disorders and the molecular etiology and classification of myeloproliferative neoplasms: From Vainchenker 1950 to Vainchenker 2005 and beyond. Acta Haematol 2015;133:36-51.
- Georgii A, Vykoupil KF, Buhr T, Choritz H, Dohler U, Kaloutsi V, et al. Chronic myeloproliferative disorders in bone marrow biopsies. Pathol Res Pract 1990;186:3-27.
- Georgii A, Buhr T, Buesche G, Kreft A, Choritz H. Classification and staging of ph-negative myeloproliferative disorders by histopathology from bone marrow biopsies. Leuk Lymphoma 1996;22 Suppl 1:15-29.
- Vannucchi AM, Antonioli E, Guglielmelli P, Pancrazzi A, Guerini V, Barosi G, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood 2008;112:844-7.
- Beer PA, Campbell PJ, Scott LM, Bench AJ, Erber WN, Bareford D, et al. MPL mutations in myeloproliferative disorders: Analysis of the PT-1 cohort. Blood 2008;112:141-9.
- Jones AV, Campbell PJ, Beer PA, Schnittger S, Vannucchi AM, Zoi K, et al. The JAK2 46/1 haplotype predisposes to MPL-mutated myeloproliferative neoplasms. Blood 2010;115:4517-23.
- Pich A, Riera L, Beggiato E, Nicolino B, Godio L, Campisi P, et al. JAK2V617F mutation and allele burden are associated with distinct clinical and morphological subtypes in patients with essential thrombocythaemia. J Clin Pathol 2012;65:953-5.
- Michiels JJ, Ten Kate F, Lam KH, Schroyens W, Berneman Z, De Raeve H. The European clinical, molecular and pathological (ECMP) criteria and the 2007/2008 revision of the world health organization (WHO) for the diagnosis, classification and staging of prefibrotic myeloproliferative neoplasms carrying the JAK2V617F mutation. Turk J Hematol 2014;31:239-54.
- Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379-90.
- Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391-405.
- Rumi E, Pietra D, Ferretti V, Klampfl T, Harutyunyan AS, Milosevic JD, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014;123:1544-51.
- Rumi E, Pietra D, Pascutto C, Guglielmelli P, Martinez-Trillos A, Casetti I, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014;124:1062-9.
- Michiels JJ, De Rave H, Valster F, Potters V, Kim Y, Kim M. Extension of 2016 world health organization (WHO) classification and a new set of clinical, laboratory, molecular and pathological (CLMP) criteria for the diagnosis of myeloproliferative neoplasms: From Dameshek Vainchenker, Green and Kralovics Eur Med J 2017a;2:72-81.
- Michiels JJ, Medinger M, De Raeve H, Schroyens W, Schelfout K, Potters V, et al. Increased erythrocyte count on top of bone marrow histology, but not by EPO level or JAK2V617F mutation load discriminates between JAK2V617F mutated essential thrombocythemia and polycythemia vera. J Hematol Thromb Dis 2015c;SI:1.
- Michiels JJ, Berneman Z, Gadisseur A, Raeve HD, Schroyens W, Potter V, et al. Myelofibrosis is a secondary event in JAK2 trilinear myeloproliferative neoplasm (MPN) and in CALR and MPL thrombocythemia: Implications for novel treatment options of prefibrotic MPN. J Hematol Thromembolic Dis 2017b;5:5.