A Rare Case of Unilateral Vogt - Koyanagi - Harada Disease
with Hypothyroidism
Anushree Gupta
Citation : Gupta A. A Rare Case of Unilateral Vogt-Koyanagi-Harada Disease with Hypothyroidism. Clin Res Ophthalmol 2018;1(2):1-4.
We present a case report of a 25-year-old Indian female who presented with severe headache, weight gain, and acute blurring of vision of the left eye. She was diagnosed as probable Vogt-Koyanagi-Harada syndrome with subclinical hypothyroidism based on laboratory tests and investigations. She was promptly started on systemic steroids along with thyroxine replacement therapy. Rapid recovery of vision with no recurrence was seen for follow-up period of 1 year.
Hypothyroidism, steroids, unilateral, Vogt-Koyanagi-Harada,Ophthalmology,asclepiusopen
INTRODUCTION
Vogt-Koyanagi-Harada disease is a systemic disorder that involves the ocular, integumentary and central nervous systems. It occurs more frequently in pigmented races, such as Asians, Hispanics, and Native Americans[1].
It is characterized by bilateral granulomatous uveitis associated with exudative retinal detachment and with extraocular manifestations. Women are affected more frequently than men mostly between the second and fifth decades of life. It is hypothesised that an autoimmune process driven by T lymphocytes is directed against a ligand associated with melanocytes or tyrosinase peptides[2].
CASE REPORT
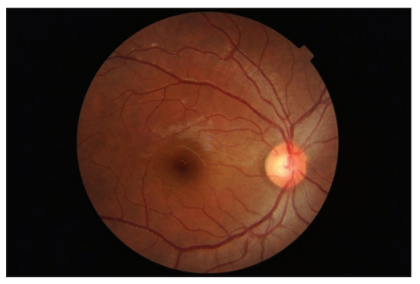
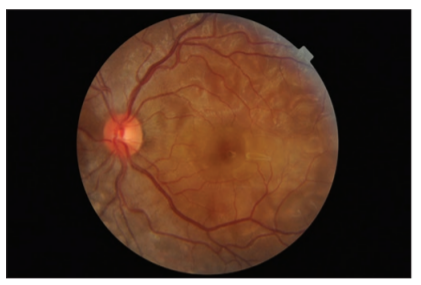
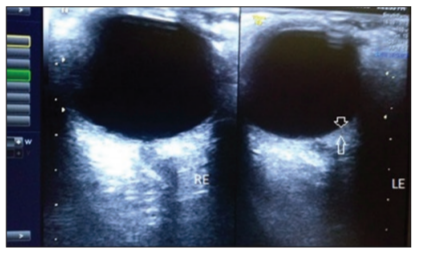
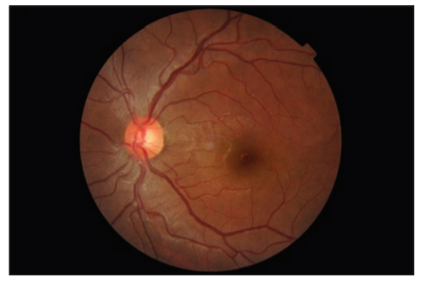
A previously healthy 25-year-old lady presented to our eye outpatient department with decreased visual acuity in the left eye for 2 weeks associated with a frontal headache. There was no history of penetrating ocular trauma or surgery. There was no history of tinnitus, vertigo, neck stiffness, or alopecia. There was a history of mild weight gain and malaise for the past few months. Rest of her medical history and systemic examination was normal. At the initial visit, the best- corrected visual acuity (BCVA) in the LE was 20/200 and in the right eye (RE) was 20/20. Intraocular pressure and pupil reaction were normal in both the eyes. Slit-lamp examination of the anterior segment showed normal findings. Right eye fundoscopy was normal, and fundus examination of the left eye showed hyperemic disc and multiple subretinal yellow lesions in the posterior pole involving the macula ( Figure 1 and 2). Ultrasound of the left eye showed thickening of the choroid (Figure 5).
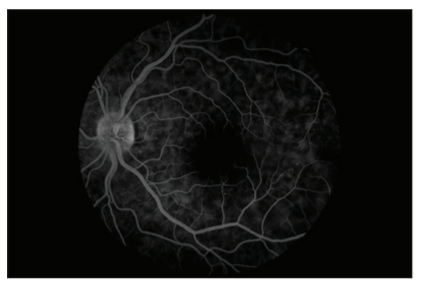
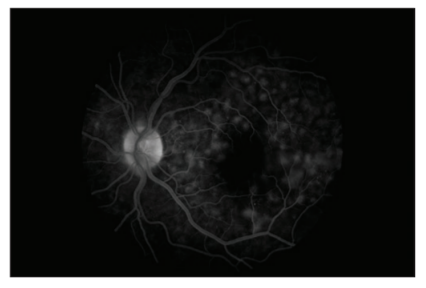
Fundus fluorescein angiography revealed multifocal areas of pinpoint leakage with progressive enlargement in the early phase and pooling of dye in the subretinal space in the late phase in the left eye ( Figure 3 and 4).
Routine blood and urine reports were normal. Laboratory tests for infectious pathologies were negative. Mantoux test and chest X-ray were normal. Neuroimaging tests such as magnetic resonance imaging brain were normal. Thyroid function test demonstrated elevated TSH and normal T4 levels on 2 separate occasions. Serum anti- Thyroglobulin antibodies were raised and positive. Anti thyroid peroxidise antibody test was negative. Clinically, patient did not have any goitre formation. Ultrasound of the neck demonstrated a heterogeneous hypoechoic thyroid gland with multiple hyperechoic septae. Lumbar puncture was not done.
Except for headache, the patient did not have any neurologic, cutaneous, or auditory manifestations.
High-steroid pulse therapy (intravenous methylprednisolone 1000 mg for 3 days) was given followed by oral steroids 60 mg once daily after breakfast along with antacid and calcium. At day 7 after starting treatment, the left BCVA increased to 20/20 and fundus examination revealed resolution of serous retinal detachments (Figure 6). Steroids were given on a tapering schedule until 12 weeks. The patient was also started on low-dose levothyroxine.
At 1 month, visual acuity was stable and decrease in choroidal thickening on ultrasound B scan was seen. Patient's visual acuity was stable for 1 year after completion of treatment, and no recurrence was seen.
DISCUSSION
We diagnosed our patient based on clinical presentation and ancillary tests as probable Vogt-Koyanagi syndrome with mild hypothyroidism. Our patient was promptly started on high-dose intravenous steroids and later oral steroids in a tapering schedule for 12 weeks. Patient was suspected to have Hashimoto thyroiditis on the basis of clinical investigations and radiological reports[3]. She was given thyroxine replacement medications.
Diagnosis of Vogt-Koyanagi-Harada (VKH) is based on the patient's clinical presentation according to the revised criteria [Table 1][4].
Although VKH is a bilateral granulomatous panuveitis, the patient had only unilateral eye involvement and presented with posterior segment disease. Cases of unilateral VKH have been reported in literature[5,6]. In a study done in South India, majority of the cases presented with posterior uveitis in the acute stage and were diagnosed as probable VKH as per the revised diagnostic criteria[5].
Ancillary testing is useful for supporting the clinical diagnosis of VKH disease. Fluorescein angiography done in acute stage shows multiple pinpoint areas of leakage at the level of the RPE. Later, they increase in size and expand into the subretinal space in areas of serous detachment leading to a large area of leakage. The optic disc can also show blurred fluorescent margins and late leakage. During the convalescent period or in chronic stage, diffuse pigmentary changes may be seen.
Lumbar puncture shows cerebrospinal fluid pleocytosis consisting of lymphocytes. The echographic manifestations include diffuse thickening of the posterior choroid, serous detachment of the retina, and vitreous opacities.
Optical coherence tomography shows subretinal fluid in exudative retinal detachments. Human leukocyte antigen typing may be used to determine genetically susceptible individuals[7].
VKH disease usually manifests as four clinical phases: Prodromal, acute uveitic, convalescent, and chronic recurrent. The prodromal stage, preceding the acute uveitic stage by a few days, may mimic a viral infection. The acute uveitic stage, which lasts for several weeks, is followed by the convalescent stage, in which the depigmentation of the tissues is more evident. Some patients may go into the chronic recurrent stage[2].
Anterior segment inflammation is treated with topical steroids. Treatment of posterior uveitis is with systemic corticosteroids. Other treatment modalities include peribulbar long-acting corticosteroids, cytotoxic agents such as cyclosporine, azathioprine, cyclophosphamide, chlorambucil, and mycophenolate mofetil[5].
Prognosis of patients who are promptly started on initial high-dose intravenous corticosteroid therapy followed by gradual tapering has been reported to have a better visual prognosis and fewer recurrences. Chronic recurrent VKH disease has been reported to be associated with more ocular complications and a worse visual outcome than initial-onset acute disease[8,9].
VKH has been reported to be associated with other autoimmune disorders such as autoimmune polyglandular syndrome type 1, hypothyroidism, diabetes mellitus, and Hashimoto's thyroiditis[10-12].
It is important to rule out other disorders by relevant clinical and laboratory investigations so that they are also managed accordingly.
The differential diagnosis of VKH includes sympathetic ophthalmia, uveal effusion syndrome, posterior scleritis, primary intraocular lymphoma, acute posterior multifocal placoid pigment epitheliopathy, acute bilateral central serous chorioretinopathy, and sarcoidosis[2].
CONCLUSION
It is important to note that VKH can rarely occur unilaterally. It can also be associated with other autoimmune disorders that should be diagnosed and appropriate treatment instituted.
REFERENCES
- Rao NA, Gupta A, Dustin L, Chee SP, Okada AA, Khairallah M, et al. Frequency of distinguishing clinical features in vogt-koyanagi-harada disease. Ophthalmology 2010;117:591-9, 599, e1.
- Albert, Jakobiec. Vogt-Koyanagi-Harada Disease (Uveomeningitic Syndrome). Principles and Practice of Ophthalmology. 3rd ed.Vol. 1. Elsevier, Saunders; 2008-10.
- Akamizu T, Amino N. Hashimoto's thyroiditis. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, et al, editors. Endotext. South Dartmouth (MA): MDText. com, Inc.; 2000. Available from: https://www.ncbi.nlm.nih. gov/books/NBK285557. [Last accessed on 2017 Jul 17].
- Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, et al. Revised diagnostic criteria for vogt-koyanagi-harada disease: Report of an international committee on nomenclature. Am J Ophthalmol 2001;131:647-52.
- Lodhi SA, Reddy JL, Peram V. Clinical spectrum and management options in vogt-koyanagi-harada disease. Clin Ophthalmol 2017;11:1399-406.
- Agrawal A, Biswas J. Unilateral vogt-koyanagi-harada disease: Report of two cases. Middle East Afr J Ophthalmol 2011;18:82-4.
- Arevalo JF. Angiography of Inflammatory Diseases in Immunocompetent and Immunocompromised Patients. In: Arevalo JF, Garcia RA, Arellanes-Garcia L, Fromow-Guerra J. Retinal Angiography and Optical Coherence Tomography. New York: Springer; 2009. p. 141-2.
- Abu El-Asrar AM, Al Tamimi M, Hemachandran S, Al-Mezaine HS, Al-Muammar A, Kangave D, et al. Prognostic factors for clinical outcomes in patients with vogt-koyanagi-harada disease treated with high-dose corticosteroids. Acta Ophthalmol 2013;91:e486-93.
- Sheu SJ, Kou HK, Chen JF. Prognostic factors for vogt-koyanagi-harada disease. J Chin Med Assoc 2003;66:148-54.
- Suzuki H, Isaka M, Suzuki S. Type 1 diabetes mellitus associated with Graves' disease and vogt-koyanagi-harada syndrome. Int Med 2008;47:1241-4.
- Jovic NS, Nesovic M, Vranjesevic DN, Ciric J, Marinkovic DC, Bonaci B. The vogt-koyanagi-harada syndrome: Association with autoimmune polyglandular syndrome Type 1. Postgrad Med J 1996;72:495-7.
- Wiesli P, Bernauer W, Furrer J. Headache and bilateral visual loss in a young hypothyroid Indian Man. J Endocrinol Invest 1999;22:141-3.