Chronic Inflammation: Prospective Prevention and/or Control by the Regulation of Nuclear Factor Kappa B with Natural Products as Dietary Supplements
Jyothirmayi Vadapalli1, Anuradha Vanam2, Noboru Motohashi3, Rao Gollapudi4*
2. Sri Venkateswara University, Tirupathi, Andhra Pradesh, India
3. Department of Medicinal Chemistry, Meiji Pharmaceutical University, 2-522-1 Noshio, Kiyose-shi, 204-8588 Tokyo, Japan
4. University of Kansas, Lawrence, Kansas-66045, USA
Citation: Vadapalli J, Vanam A, Motohashi N, Gollapudi R. Chronic Inflammation: Prospective Prevention and/or Control by the Regulation of Nuclear Factor Kappa B with Natural Products as Dietary Supplements. J Community Prev Med 2018;1(1):1-12.
Nuclear factor kappa B (NF-kB) refers to a group of five structurally related and conserved proteins in mammals including Re1A/p65, Rel/cRel, Re1B, NF-kB 1/p50, and NF-kB 2/p52. A major regulation of NF-kB activity is through the control of NF-kB migration between nucleus and cytoplasm. Deregulation of NF-kB activation pathway at any stage results in continuous NF-kB activation and chronic inflammation and eventually cancer. By trapping NF-kb in cytoplasm, IKB prevents NF-kB/ deoxyribonucleic acid complex formation in the nucleus. A chronic inflammation is related to the integral activation of NF-kB because of an imbalance in inflammatory signaling network including defective anti-inflammatory mechanism and tenacious infection with pathogens. Chronic inflammation results in terminal damage to blood vessels, cells, and organs causing multiple health problems such as diabetes, cancers, asthma, cardiovascular diseases, psoriasis, and neurodegenerative diseases as well as joint pains. A number of small molecules from natural resources including fruits, vegetables, and animals could inhibit or prevent chronic inflammation and its related ailments.
Angiogenesis, apigenin, astaxanthin, asthma, cancers, capsaicin, cardiovascular diseases
INTRODUCTION
Inflammation is body inbuilt defense mechanism as a response to injuries. Chronic inflammation is a prolonged inflammation resulting in noticeable terminal damage to blood vessels, cells, and organs causing multiple health problems such as diabetes, cancers, asthma, cardiovascular diseases, psoriasis, and neurodegenerative diseases as well as joint pains.
Angiogenesis and inflammation had been linked to the imperative basis of multiple chronic inflammatory disorders including Crohn's disease, lupus, inflammatory bowel disease, sepsis, gastritis, atherosclerosis, rheumatoid arthritis, psoriasis, diabetes, cancer, and metastasis. Environmental and habitual factors like pollution, lack of exercise, diet, stress, obesity, smoking, oral health and excessive alcohol consumption might lead to chronic inflammation. As a result, white blood cells form groups without any purpose and ultimately attack cells and tissues of internal organs. In general, chronic inflammation did not reveal any symptoms but only could be detected by elevated C-reactive protein levels (CRP)[1-3].
CHRONIC INFLAMMATION-ASSOCIATED AILMENTS
In cardiovascular diseases, cholesterol gets deposited in the blood vessel lining acting as an insult leading to systematic inflammation, resulting in blockages and blood clots causing heart attacks. Hence, people with chronic inflammation from an autoimmune disorder might be at greater risk for heart disease. In addition, bacteria from gum disease reached blood vessels and heart causing inflammation that elevated the chance of heart attack. The increased levels of some nuclear factor kappa B (NF-ΚB) activators such as osteoprotegerin were linked to cardiac disease-related mortalities[4,5].
In diabetes, nuclear factor kappa-light-chain-enhancer of activated beta cells (NF-ΚB) was activated by multiple pro-inflammatory cytokines for normal survival and death of β-cells. β-cells were destroyed with the activation of inducible nitric oxide synthase (iNOS) gene expression and successive formation of NO. Interleukin-1 β (IL-1β)-induced NF-ΚB activation resulted in apoptosis of β-cells in the pancreas, in type-1 diabetes, whereas, in type-2 diabetes, activated NF-ΚB induced apoptosis and insulin resistance. NF-ΚB was upregulated with interactions of advanced glycation end products and their receptor advanced glycation end products. Uninterrupted activation of NF-ΚB stimulated a systemic inflammation, a contributory factor for the development of multiple diabetic ailments such as diabetic cardiomyopathy, retinopathy, nephropathy, and neuropathy, suggesting a need for NF-ΚB-based therapeutic approach[6].
Chronic inflammation in lungs was associated with various problems such as chronic obstructive pulmonary disease (COPD) and asthma. Inflammation of the lungs resulted in fluid accumulation, thereby narrowing the airways making breathing arduous. COPD developed as a chronic and significant inflammatory response to inhaled irritants. COPD was a third most common cause of death in the United States[7,8].
Individuals with high aggressive behavior displayed elevated inflammatory cytokine levels and deregulated immune responses including slower wound healing. Cytokines produce sickness behaviors including reduced activity, food intake, and social interaction together with increased sleep and anhedonia. Higher levels of inflammatory markers such as tumor necrosis factor alpha (TNF-a), CRP and IL-6 were found in people with intermittent explosive disorder appeared to be related to the aggressive behavioral aspect in humans. Increased NF-ΚB inflammatory signaling included the elevated expression of pro-inflammatory cytokines[9,10].
Chronic inflammation was linked to increased bone loss and lack of bone growth where cytokines in the blood interfered with bone renovation, a process in which damaged old bones were replaced with the new. In addition, inflammation in gut decreased the absorption of nutrients such as calcium and vitamin D which were essential to bone health[11].
When immune cells infiltrated the tumor in an inflammatory response, tumor grows utilizing those immune cell nutrients and oxygen instead of getting destroyed. Gene mutations occurred as a result of chronic inflammatory response which triggered the loss of proteins involved in deoxyribonucleic acid (DNA) repair. Occasionally, chronic inflammation might serve as a precursor to certain cancers associated with DNA damage which could lead to cancer. People with chronic inflammatory bowel diseases, ulcerative colitis, and Crohn's disease were at an increased risk of colon cancer[12].
It has long been assumed that NF-ΚB signaling played an important role in chronic inflammation-associated malignancies and other inflammation-associated disorders although genetic evidence for this hypothesis had been lacking. However, recent studies suggested the vitality of NF-ΚB activation in tumor cell survival, angiogenesis, and metastatic potential. NF-ΚB activation of tumor-associated leukocytes and macrophages, in particular, resulted in tumorogenesis through the upregulation of tumor-promoting pro-inflammatory proteins, emphasizing the importance of NF-ΚB inhibitors as immunotherapeutic agents to reduce or prevent chronic inflammation. Therefore, it was obvious that NF-ΚB inhibitors might inhibit or prevent chronic inflammation-related tumorogenesis as well as angiogenesis and metastasis[13,14].
CELLULAR MECHANISMS OF NF-ΚB ASSOCIATED INFLAMMATION
A number of inflammatory mechanisms occurred by the activation of inflammatory factor, NF-ΚB, a protein complex controlled transcription of DNA, cytokines production, and cell survival. In addition, NF-ΚB was involved in cellular responses to inflammation stimuli caused by cytokines, heavy metals, ultraviolet irradiation free radicles, bacterial or viral antigens, and oxidized low-density lipids including stress. The unwanted inflammation process could be reduced by switching off NF-ΚB actions thereby minimizing damage to cells, tissues, and organs. Recognized inducers of NF-ΚB activity were TNF interleukin 1-β (IL-1β), reactive oxygen species (ROS), osteoprotegerin, isoproterenol, bacterial lipopolysaccharides (LPS), cocaine, and ionizing radiation. The recognized activity of NF-ΚB was associated with the regulation of inflammatory responses as well as activation, differentiation, and effective function of inflammatory T-cells. The increased levels of TNF-a regulated protein kinase B (AKT)/mammalian target of rapamycin pathway were essential for the management of skeletal muscle hypertrophy. NF-ΚB pathway regulated pro-inflammatory cytokine production and leucocyte recruitment which were primary contributors to inflammatory response. NF-ΚB reduction contributed to the control of multiple inflammatory mechanisms thereby diminishes enormity and duration of inflammatory response.[15,16] [Figure 1].
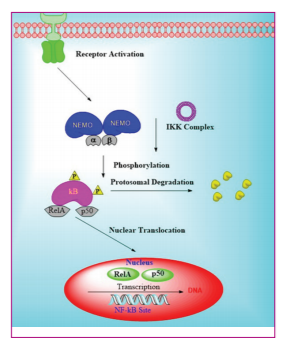
NF-ΚB was a central regulator of distinctive immune response. Usually, NF-ΚB was activated as host protective mechanism, and long-term activation of NF-ΚB was tumorigenic in nature. Furthermore, NF-ΚB recurrent activation obstructed the activities of inflammatory mediator, resulting in tumor progression. Multiple small molecules from natural or synthetic origin targeted different signaling pathways including NF-ΚB pathway and p53 protein (p53), thereby establishing a prominent change in cancer treatment and management. The anticancer activity of various NF-ΚB inhibitors was partly due to their capacity to induce p53 in cancer cell[17].
Selective natural products were inhibitors of NF-ΚB signaling by intercalating to the enhancer sequences of heavy chain of immunoglobulin and gene kappa light chain. NF-ΚB was a group of interrelated transcription factors including five genes: NF-ΚB1 (p50/p105), NF-ΚB2 (p52/p100), RelA (p65), c-Rel, and RelB. In cancers, inflammatory stimuli controlled the mechanisms of gene expression. In this process, the cells ceased to relate their existence with underlying mechanisms, coordinating their phenotype and functions. NF-ΚB activation was triggered by two separate pathways such as canonical and non-canonical (alternative) pathways. The canonical pathway was activated by toll-like receptors and pro-inflammatory cytokines (TNFa and IL-1), directing the activation of Re1A that controlled the expression of pro-inflammatory and cell survival genes. The alternative NF-ΚB pathway was activated by lymphotoxin β (L? β), CD40 ligand, B-cell activating factor belonging to the TNF family (BAFF), and receptor activator of NF-ΚB ligand (RANKL) resulting in activation of RelB/p52 complexes. Alternative pathway activation regulated genes that were required for lymph-organogenesis and B-cell activation. A variety of cytokines, growth factors, and kinases involved in signaling pathways triggered the activation of NF-ΚB, key protein, a major therapeutic target for drug discovery in cancer inflammation and progression[18-21]. NF-ΚB signaling system had been established to be a mediator of inducible and tissue-specific gene control. Nonetheless, NF-ΚB/REL complexes contained homoor heterodimers of the proteins of NF-ΚB and Rel families. The Rel family included RelA p65, c-Rel, and RelB proteins, whereas NF-ΚB family comprised p50 (p105) and p52 proteins (p100). Usually, NF-ΚB complexes were localized in cytoplasm by binding to inhibitory IkB proteins. (IkBα a, IkBβ, IkBγ, IkBε and Bcl3). Phosphorylation of IkB proteins transpired through activation of either external or internal signals. Later, they were ubiquitinated and destroyed in proteasomes. IkB protein release from Rel homology domain of Rel protein disclosed nuclear localization sequence domain. Furthermore, NF-ΚB-complex migrated into nucleus and activated the transcription of genes including inflammatory genes. There were variations in the activation of NF-ΚB-complex signaling pathways upstream. Furthermore, there were alterations in transactivation ability of NF-ΚB-complexes at the transcription level. The protein kinases that phosphorylated IkB proteins were I-kappa B kinases (IKKs) (IkB kinases a and β). NF-ΚB-signaling pathway is NF-ΚB-essential modulator and regulatory subunit of IKK complex. IKKs were controlled by various interacting proteins by connecting IKK complex to canonical pathway thereby regulating the activation of IKK. The other non-canonical NF-ΚB pathway was activated by NF-ΚB-inducing kinase (NIK) which facilitated signals from CD40, lymphotoxin, and BAFF/BLys receptors. This pathway was IKK dependent and however, IΚB-independent. Furthermore, non-canonical NF-ΚB pathway regulated NF-ΚB-activation through p100 (NF-ΚB-p52) subunit handling.. IKKa/IKKβ was a junction for NF-ΚB-mediated inflammatory signaling. Furthermore, several cytokine receptors were connected to NF-ΚB-signaling to increase and enumerate the inflammatory responses. NF-ΚB-system was a cytoplasmic sensor that responded to immune assaults as well as to various external and internal hazard signals such as hypoxia, oxidative, and genotoxic stress. NF-ΚB-signaling signified the connection between inflammation and cancer. The genes responsible for inflammation contributing to the activation of NF-ΚB-signaling were important targets for drug discovery[22-25] [Figure 2].
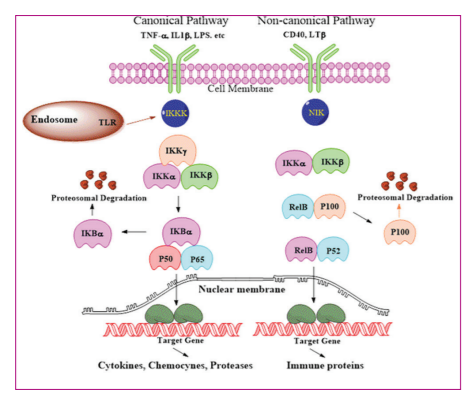
Natural products as anti-inflammatory activators through the suppression of NF-ΚB signaling.
Several herbal remedies proved to be potent drugs against various NF-ΚB - associated inflammatory ailments and cancer. Selective inhibition of IKKβ, a mediator of innate immune responses and cancer, proved to be a proper strategy to block NF-ΚB - signaling. Various plant-derived products had been established as possible inhibitors of NF-ΚB-pathway including lignans, polyphenols, and terpenoids[26]. Various natural products inhibited inducible as well as constitutively active NF-ΚB-activities, and some of these compounds with specificity toward IKK or IKKK, IΚBa stability, p65 translocation, and DNA binding in NF-ΚB-activation pathway have been reported[27].
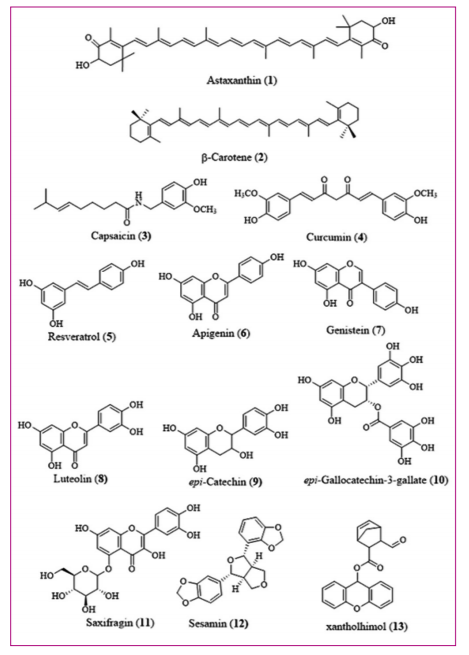
Astaxanthin, (1) a xanthophyll mostly present in salmon, shrimp, and crab, blocked the activation of IKKa kinase and IΚBa protein degradation as well as nuclear movement of NF-ΚB-p65 subunit in addition to inflammatory NF-ΚB-dependent gene expression, thereby reducing inflammation through its antioxidant activity[28,29]. β-carotene (2) suppressed LPS-induced NF-ΚB-signaling and expression of inflammatory genes by blocking the degradation of IΚBa protein, nuclear migration of p65 protein, DNA binding of NF-ΚB-complex, LPS-induced expression iNOS, cyclooxygenase-2 (COX-2), TNF-a, and IL-1β expression[30].
Capsaicin (3), a known inhibitor of NF-ΚB, from chili peppers (Capsicum species) prevented IΚBa degradation and nuclear translocation of p65. Moreover, capsaicin (3) prevented NF-ΚB-activity by blocking the degradation and phosphorylation of IΚBa. Capsaicin (3) inhibited production of LPS-induced prostaglandin E2 (PGE2) and curbed COX-2 enzyme activity as well as the expression of iNOS protein. Capsaicin (3) entirely blocked LPS-induced disappearance of IkB-a and inactivated NF-ΚB. Capsaicin (3) inhibited constitutive as well as IL-1β-induced and TNF-a-induced IL-8 expression[31,32]. Curcumin (4), a major constituent of Curcuma longa (turmeric) rhizomes, inhibited IKK, pro-inflammatory TNF-2a, COX-1, COX-2, and p53 activation by inhibiting mouse double minute 2 homologue and regulating other signaling pathways. Curcumin (4) inhibited the expression of COX-2 gene induced by phorbol 12-m/ restate 13-acetate and TNF-a or fecapentaene-12 in human colon epithelial cells. Curcumin (4) wedged tumor promoter-mediated NF-ΚB-transactivation through inhibition of NIK/ IKK signaling complex. Furthermore, curcumin (4) suppressed IKK and inhibited constitutive and inducible NF-ΚB-activation as well as strengthened TNF-a-apoptosis. Curcumin (4) curbed Ras/mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase/AKT signaling pathways that were involved in the activation of NF-ΚB[33-36]. Resveratrol (5) obstructed NF-ΚB/p65 and p53transcriptional functions by the deacetylation of specific residues. Resveratrol (5) treatment increased chromatin-associated sirtuin 1 (SIRT1), cellular inhibitor of apoptosis 2 promoter regions in the cells. This effect correlated with the loss of NF-ΚB-regulated gene expression and cell sensitivity to TNFa induced apoptosis suggesting that SIRT1 activity increased apoptosis in response to TNFa with decatalyse capacity to inhibit the transactivation capacity of RelA/p65 protein[37-40]. Apigenin (6) present in parsley, thyme, and peppermint intercepted p65 phosphorylation by inhibiting IKK functions. In addition, apigenin (6) suppressed NF-ΚB-translocation to nucleus which resulted in the inhibition of IΚBa degradation and phosphorylation. Apigenin (6) regulated NF-ΚB-activity through hypophosphorylation of Ser 536 in P65 subunit, in non-canonical pathway. Furthermore, apigenin (6) inactivated IKK complex stimulated by LPS. In addition, apigenin (6) inhibited LPS-induced TNF in vivo. Besides, apigenin (6) inhibited mortality induced by lethal doses of LPS suggesting a molecular mechanism of apigenin (6) in inflammation suppression and modulation of immune response[41]. Apigenin (6) strengthened activation-induced cell death by inhibiting NF-ΚB-activation and supressing NF-ΚB-regulated antiapoptotic molecules (cFLIP, Bcl-x (L), Mcl-1, XIAP, and IAP) and supressed NF-ΚB-translocation to nucleus. Moreover, apigenin (6) inhibited IΚBa phosphorylation and degradation as a response to T-cell receptor (TCR) stimulation in reactivated peripheral blood CD4-positive T-lymphocyte (CD4 T cells). Besides, apigenin (6) suppressed the expression of COX-2 protein in activated human T-cells[42].Genistein (7) from soybeans and fava beans obstructed the activation of NF-ΚB-and degradation of IΚBa as well as inhibited NF-ΚB-signaling through AKT pathway. Genistein (7) treatment of human myeloid cells, T-cells, and epithelial cells completely suppressed TNF-induced NF-ΚB-activation correlated with protein tyrosine kinase activity. In addition, genistein (7) inhibited the activation of NF-ΚB-and AKT signaling pathways which maintained the balance between cell survival and programmed cell death (apoptosis), angiogenesis, and metastasis[43,44]. Luteolin (8) present in celery, broccoli, green pepper, parsley, and thyme prevented NF-ΚB-activity through the accumulation of ROS. Luteolin (8) markedly controlled NF-ΚB-activation while potentiated c-jun amino-terminal kinase c-jun N-terminal kinase (JNK) to increase apoptosis induced by TNF in lung cancer cells. Furthermore, luteolin (8) induced an early phase of ROS segregation through suppression of cellular superoxide dismutase (SOD) activity. Therefore, accumulating ROS induced by luteolin (8) played an important role in the suppression of NF-ΚB-and potentiation of JNK to sensitize lung cancer cells to go through TNF-induced apoptosis[45]. Epi-catechin (9) an important constituent of green tea, coco, and grapes stalled the constitutive NF-ΚB-activity by obstructing p65 nuclear translocation and inhibited NF-ΚB DNA-binding activity. Epi-catechin (9) inhibited NF-ΚB-DNA binding by preventing NF-ΚB-as well as NF-ΚB-dependent gene expression in L-428 and KM-H2 cells[46]. Epi-gallocatechin-3-gallate (10), a constituent of green tea, reduced IKK activation, IΚBa degradation, NF-ΚB-activation, and phosphorylation of p65 subunit of NF-ΚB-and prevented nuclear translocation of p65. Epi-gallocatechin-3-gallate (10) reduced IL-1 β-mediated IL-1β receptor-associated kinase (IRAK) degradation and the subsequent downstream of signaling episodes, IKK activation, IΚBa degradation and NF-ΚB-activation. Besides Epi-gallocatechin-3-gallate (10) curbed phosphorylation of p65 subunit of NF-ΚB-which was evident by the inhibition of IL-8 gene expression. Epi-gallocatechin-3-gallate (10) inhibited prototype tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced DNA binding of NF-ΚB-and cyclic adenosine 3',5'-monophosphate response element binding protein (CREB) in mouse skin. Furthermore, epi-gallocatechin-3-gallate (10) repressed TPA-induced phosphorylation and the consequent degradation of IΚBa and simultaneously restricted nuclear translocation of p65. Epi-gallocatechin-3-gallate (10) inhibited TPA-induced DNA binding of NF-ΚB-and CREB by blocking activation of p38 MAPK. suggesting a molecular basis of COX-2 inhibition by epi-gallocatechin-3-gallate (10) in mouse skin. Epi-gallocatechin-3-gallate (10) inhibited build-up of LPS-induced IL-12p40, IL-6, monocyte chemoattractant protein-1, intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion protein, and mRNA in bone marrow-derived macrophages. Moreover, epi-gallocatechin-3-gallate (10) restricted LPS-induced IΚBa degradation as well as RelA nuclear translocation. Consequently, epi-gallocatechin-3-gallate (10) could prevent LPS-induced pro-inflammatory gene expression through the restriction of NF-ΚB-and MAPK signaling pathways[47-49] Saxifragin (11) (quercetin 5-glycoside) is widely distributed in plants as well as insects and displayed peroxynitrite-scavenging effects. Saxifragin
(11) suppressed the production of NO and PGE2 in LPS-activated RAW 264.7 macrophages by suppressing the level of protein and mRNA expression of iNOS and COX-2, respectively. In addition, saxifragin (11) inhibited the mRNA expression of pro-inflammatory cytokines comprising TNF-a, IL-6, and IL-1β. The inhibitory effects of saxifragin (11) on NF-ΚB were a result of activation of caspase-1 and phosphorylation of Jun-N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK). Moreover, pretreatment with saxifragin (11) increased the survival rate of mice with LPS-induced septic death. Thus, saxifragin (11) displayed anti-inflammatory activity through the inhibition of NF-ΚB, caspase-1, and MAPK activation [50] Sesamin (12) from the bark of Fagara species and sesame oil prevented TNF-induced IKK activation. IΚBa degradation and phosphorylation; down-regulated constitutive and inducible NF-ΚB-activation and suppressed P65 phosphorylation and nuclear translocation. Sesamin (12) assisted in the prevention of hyperlipidemia, hypertension, and carcinogenesis and inhibited the proliferation of wide variety of tumor cells including leukemia, cancers of colon, pancreas, lung, prostate, and breast as well as multiple myeloma. In addition, sesamin (12) increased TNFa induced apoptosis associated with suppression of gene products related to cell survival (Bcl-2 (B-cell leukemia/lymphoma 2 protein Bcl-2)), proliferation bcl-1 proto-oncogene product (cyclin D1)), inflammation (COX-2), invasion (matrix metalloproteinases-9 [MMP-9] and ICAM-1), and angiogenesis. Sesamin (12) reduced constitutive and inducible NF-ΚB-activation which was initiated by multiple inflammatory stimuli and carcinogens. Furthermore, sesamin (12) reduced the degradation of IΚBa by supressing phosphorylation of IΚBa there by inhibiting the activation of clampdown of p65 phosphorylation and nuclear translocation as well as NF-ΚB-mediated reporter gene transcription. Moreover, sesamin (12) markedly reduced LPS-stimulated IL-6 mRNA and protein in microglia cells (BV-2). Sesamin (12) decreased LPS-activated p38 MAPK and NF-ΚB-activation. Furthermore, SB203580 (inhibitor of p38 MAP kinase) inhibited LPS-induced IL-6 production [51],[52]. Xantholhumol (13) (Hops) controlled T cell-mediated immune responses by inhibiting NF-ΚB-transcription factor through the suppression of phosphorylation of IΚBa (inhibitor of NF-ΚB) [Figure 3] [53]
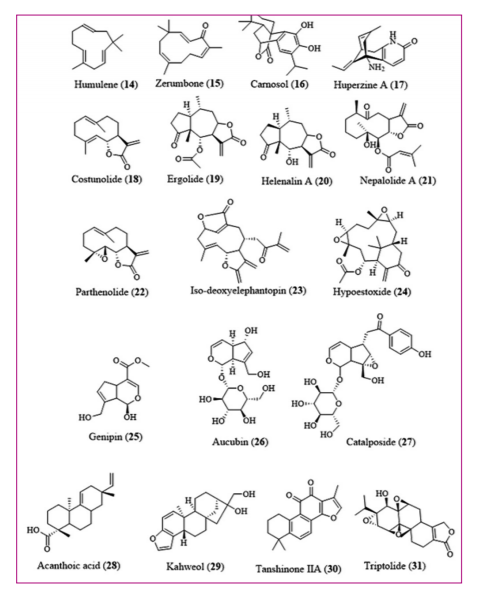
A number of monoterpenoids, sesquiterpenoids, and diterpenoids displayed anti-inflammatory activity by intercepting NF-ΚB-signaling pathways. Humulene (14) present in Humulus lupulus (hops) significantly reduced LPS-induced NF-ΚB-activation as well as inflammatory response.
However, humulene (14) did not alter the activation of ERK, p38, and JNK [54] Zerumbone (15), a potent anti-inflammatory and anticancer agent isolated from Zingiber zerumbet (ginger), curbed the function of IKK complex as a result of reduced protein phosphorylation and degradation of IΚBa) proteins, consequently resulting in a decrease in nuclear translocation of NF-ΚB-complex and gene expression [55,56] Carnosol (16) from Rosmarinus officinalis (Rosemary) curbed the activation of NF-ΚB-system through inhibiting IΚBa phosphorylation and reducing the expression of iNOS and NO production. Metastasis was suppressed by carnosol (16) through the blockade of MMP-9 expression with the downregulation of NF-ΚB-and c-Jun protein-mediated signaling as a result of its antioxidant capacity [57,58], Inflammatory responses were suppressed by huperzine A (17) from Huperzia serrata with the restriction of NF-ΚB-signaling [59]. Costunolide (18) from Saussurea lappa (costus root oil) inhibited phosphorylation of IkB proteins, resulting in nuclear localization of NF-ΚB-complex. Furthermore, costunolide (18) inhibited LPS-induced inflammatory signaling pathway by curbing NF-ΚB-activation and downstream gene expression [60]. Ergolide (19) from Inula britannica (British yellowhead and Meadow fleabane) constrained NF-ΚB-activation in LPS-stimulated RAW 264.7 macrophages through inhibition of nuclear translocation of NF-ΚB-complex and degradation of IkB protein [61]. Helenalin A (20) from Arnica montana (wolf's bane, Leopard's bane, Mountain tobacco, and Mountain arnica) and Arnica chamissonis (Chamisso arnica) inhibited NF-ΚB-signaling through alkylation of p65 subunit of NF-ΚB complex, thereby inhibiting the complex DNA binding and transcription of NF-ΚB-dependent genes [62]. Nepalolide A (21) from Carpesium nepalense displayed anti-inflammatory activity through the inhibition of LPS- and cytokine- induced activation of NF-ΚB-signaling in C6 glioma cells. The suppression of NF-ΚB-signaling occurred due to the inhibition of IkB protein phosphorylation in stimulated cells [63]. Parthenolide (22) of Tanacetum parthenium (feverfew) repressed the activity of IKKβ, a kinase subunit that played a vital function in cytokine-mediated signaling. Mutation of cysteine 179 in activation loop of IKKβ resulted in elimination of IKKβ binding sensitivity to parthenolide (22). Furthermore, anti-inflammatory activity of parthenolide (22) was facilitated through a-methylene ?-lactone moiety present in other sesquiterpene lactones. Parthenolide (22) alkylated cysteine-38 in p65 subunit of NF-ΚB-and inhibited DNA binding of NF-ΚB-complex [64]. Iso-deoxyelephantopin (23) from Elephantopus scaber inhibited osteoclastogenesis by suppressing NF-ΚB-activation and potentiated apoptosis. Furthermore, iso-deoxyelephantopin (23) reduced cytokine-induced NF-ΚB-activation by suppressing IKK complex activity [65]. Hypoestoxide (24) from Hypoestes rosea inhibited IKKβ activation and inflammatory responses including colorectal cancer [66,67]. Genipin (25), from Gardenia fruit extract, displayed anti-inflammatory activity by inhibiting the expression of iNOS and NO production in RAW 264.7 macrophages. Genipin (25) restricted the degradation of IkBb protein that led to inhibition of NF-ΚB-signaling [68]. Aucubin (26), an iridoid glycoside from Rehmannia glutinosa, exhibited its anti-inflammatory activity by inhibiting the degradation of IΚBa protein and prevented the nuclear translocation of p65 subunit of NF-ΚB-complex. In addition, aucubin (26) acted as anti-inflammatory agent protecting against hepatotoxicity. Besides, aucubin (26) exhibited antitumor activity [69,70]. Acanthoic acid (27) from the bark of Acanthopanax koreanum curbed LPS-induced activation of IΚBa phosphorylation and nuclear DNA binding of NF-ΚB-complex in addition to the reduction in LPS-induced cytokine synthesis and pro-inflammatory response [71]. Kahweol (28) present in Coffea arabica (coffee beans) suppressed NF-ΚB-related transcriptional activation through inhibition of nuclear DNA binding of NF-ΚB-complex, IKK activity and prevented degradation of IkB proteins [72,73]. Catalposide
(29) from Catalpa ovata (yellow catalpa. Chinese catalpa) curbed the activation of NF-ΚB, IΚBa protein degradation and translocation of P65 sub-unit to the nuclei. Furthermore, catalposide (26) decreased TNF-a induced p38 and ERK phosphorylation through up-regulation of cytokine signaling thereby reducing intestinal inflammation [74]. Tanshinone IIA
(30) from Salvia miltiorrhiza suppressed NF-ΚB-signaling and inhibited IKKa/β and NIK activation, consequently inhibiting the phosphorylation of IΚBa protein as well as nuclear migration NF-ΚB-complex [75]. Triptolide (31) from Tripterygium wilfordii inhibited the phosphorylation of NF-ΚB-complex into nuclei and ultimately DNA binding of the complex. Furthermore, triptolide 32 curbed NF-ΚB-signaling in T-lymphocytes by upregulating IΚBa protein expression [76,77] [Figure 4].
Several triterpenoids and their glycosides displayed anti-inflammatory activity by interfering with NF-ΚB signaling pathway reducing inflammation. Betulinic acid (32) (pentacyclic triterpenoid), naturally occurring and widely distributed in plants such as Ancistrocladus heyneanus, Diospyros leucomelas, Prunella vulgaris (common selfheal), Pseudocydonia sinensis (Chinese quince), Pulsatilla chinensis, R. officinalis (rosemary), Syzygium formosanum (jambul), Tetracera boiviniana, Triphyophyllum peltatum, and Ziziphus mauritiana displayed anti-inflammatory activity by the suppression of IKKa activation which was initiated by certain typical NF-ΚB-activators followed by the downregulation of NF-ΚB-dependent gene expression [78]. Glycyrrhizin (33) from Glycyrrhiza glabra (liquorice) with the help of glycyrrhizic acid inhibited NF-ΚB-signaling. The calcium-mediated activation of NF-ΚB-system was blocked by glycyrrhizic acid. However, excessive use of liquorice could result in hypertension [79]. Lupeol (34) present in various fruits, vegetables, and several herbs inhibited NF-ΚB-signaling including phosphorylation of IΚBa protein, DNA binding of NF-ΚB-complex as well as NF-ΚB-related gene activity. Furthermore, lupeol (30) inhibited various signaling pathways such as AKT-dependent pathways, reducing the inflammation [80-82]. Acetyl-11-keto-β-boswellic acid (35) from Boswellia serrata curbed constitutively activated NF-ΚB-signaling by inhibiting IKK activity. Psoriasis vulgaris is a chronic inflammatory skin disease involving cytokines and activated cellular immune system where psoriatic skin lesions display potent activation of NF-ΚB, mainly confined to dermal macrophages. Severe psoriasis lesion topical treatment with IKKβ inhibitor and acetyl-11-keto-β-boswellic acid (35) resulted in profound suppression of TNFa production of macrophages [83]. Celastrol (36) from Celastrus, tripterygium wilfordii, C. orbiculatus, and C. regelii intercepted the systolic I-Ba degradation and nuclear translocation of RelA and blocked IKK function together with IKKβ activity. Celastrol (36) inhibited numerous stimuli-induced NF-ΚB-regulated gene expression and DNA-binding of NF-ΚB-without affecting DNA-binding activity of activator protein-1 (AP-1). Celastrol (36) pre-incubation entirely blocked LPS-, TNF-a-, or phorbol 12-myristate 13-acetate (PMA)-induced degradation and phosphorylation of IΚBa. Celastrol (36) primarily inhibited IKK and constitutively active IKKβ activities. Furthermore, NF-ΚB-activation was suppressed by celasterol (36) through targeting cysteine 179 in IKK. Celastrol (36) prevented LPS-induced messenger ribonucleic acid (mRNA) expression iNOS and TNF--a as well as TNFa-induced antiapoptotic protein-BfI-1/A1 BfI-1/A1 expression. Celastrol (36) suppressed proliferation, invasion, and angiogenesis through the induction of apoptotic factors and reducing constitutive NF-ΚB-activity [27,84]. Ursolic acid (37), a natural pentacyclic triterpenoid carboxylic acid present in wide variety of plants, including apples, basil, bilberries, cranberries, peppermint, rosemary, and oregano inhibited the activation of NF-ΚB-signaling initiated by different carcinogenic factors in various cell lines. Ursolic acid (37) restricted IΚBa kinase activation, IkBa protein phosphorylation and degradation, p65 migration to nucleus and DNA binding of NF-ΚB-complex including NF-ΚB-related gene expression [85]. Escin (38), a constituent of Aesculus hippocastanum (horse chestnut), restricted TNF-induced IKK activation as well as I- Ba degradation and phosphorylation. Escin (38) strengthened TNF-induced apoptosis and inhibited tumor cell invasion. This process was associated with the downregulation of B-cell leukemia/lymphoma 2 protein (Bcl-2) cellular inhibitor of apoptosis bcl-1 proto-oncogene product (cyclin D1), COX-2, intercellular adhesion molecule-1, MMP-9, and vascular endothelial growth factor (VEGF) which were regulated by t NF-ΚB-activation. Accordingly, escin (38) inhibited the activation of NF-ΚB-through IKK inhibition resulting in sensitization of cells to cytokines and chemotherapeutic agents [86]. Saikosaponin D (39) from Bupleurum curbed NF-ΚB-signaling along with T-cell activation and apoptosis of cancer cells including inflammation. Saikosaponin D (39) blocked the phosphorylation of IΚBa protein and increased protein level of inhibitory IΚBa protein. In addition, saikosaponin D (39) constrained Jun-N-terminal kinase (JNK. c-jun amino-terminal kinase) signaling through upstream regulation of IKK and NF-ΚB-complexes [Figure 5] [87].
Avicin G (40), a triterpenoid glycoside from Acacia victoria (gundabluie and bardi bush), inhibited DNA binding of NF- ΚB-complex and expression of N-kB-dependent genes, resulting in the reduction of inflammation [Figure 5] [88]. Ginsenosides, a mixture of saponins of Panax species (ginseng) prevented the activation of IKKa kinase and phosphorylation and degradation of IΚBa protein thereby inhibiting NF-ΚB - signaling either directly or indirectly. Ginsenosides, the main components of Panax ginseng, were well known for their anti- inflammatory and antiproliferative activities. Ginsenoside Rb 1 (42) was converted by intestinal bacteria to compound K (41) . This metabolite showed a significant inhibitory effect on TNF-a- induced expression of intercellular adhesion molecule-1 in human astroglial cells. Pretreatment with compound K (41) suppressed TNF-a-induced phosphorylation of ??βa kinase and the subsequent phosphorylation and degradation of IkBa. In addition, the treatment inhibited TNF-a-induced phosphorylation of mitogen-activated protein kinase kinase 4 and subsequent activation of JNK-activating protein kinase 1 (JNK-AP-1) pathway, suggesting that ginsenoside metabolite compound K (41) displayed anti-inflammatory effect through the inhibition of both NF-ΚB-and JNK pathways in a cell-specific manner [89]. Pregnane X receptor (PXR) activation displayed anti-inflammatory effects by blocking NF-ΚB. However, overactivation of PXR might disturb the homeostasis of multiple enzymes and transporters. Ginsenosides curbed NF-ΚB activation and reinstated the expression of PXR target genes in TNF-a-stimulated LS174T cells. In addition, ginsenosides restrained NF-ΚB activation in a PXR-dependent manner and increased interaction between PXR and NF-ΚB p65 subunit and thus decreased the nuclear translocation of p65. Ginsenoside Rb1 and compound K (41) were major bioactive compounds in controlling PXR/NF-ΚB signaling pathway. Ginsenosides attenuated dextran sulfate sodium-induced experimental colitis, associated with restored PXR/NF-ΚB signaling, suggesting that ginsenosides might cause anti-inflammatory effects by targeting PXR/NF-ΚB interaction without disrupting PXR function [Figure 6] [90].
In another study, compound Rb1 and its metabolite compound K (41) could inhibit colitis injury. Compound K (41) lessened colitis histopathology injury and improved myeloperoxidase activity. Furthermore, compound K (41) reduced pro-inflammatory cytokines production, such as IL-6, IL-1β, TNF-a, and elevated anti-inflammatory cytokine IL-10 production. Compound K (41) inhibited NF-ΚB p65 nuclear translocation, downregulated p-IΚBa, and upregulated IΚBa, suggesting that compound K (41) suppressed the activation of NF-ΚB pathway in the progression of colitis [Figure 6] [91].
Several secondary metabolites regulated the defect in inflammatory pathways by suppressing NF-ΚB-activity with high selectivity. Epidemiological data suggested that intake of small amounts of polyphenols from foods and beverages exerts a strong effect in the reduction of inflammation and chronic diseases. It had been widely acknowledged that many plant-derived compounds exhibited significant anti-inflammatory effects. These naturally occurring compounds displayed anti-inflammatory properties by their actions with the modulation of cytokines and associated intracellular signaling pathways [92,93].
CONCLUSION
Plant and animal-derived constituents could prevent or inhibit NF-ΚB-signaling system, displaying therapeutic effects against inflammatory ailments including cancer. NF-ΚB, a key regulator of internal immune response, is activated as a host protection. However, chronic inflammation is tumorigenic and blocking the activities by inflammatory mediators suppresses tumor regression and its aggressiveness. Two separate pathways for NF-ΚB-activation include canonical pathway and non-canonical (alternative) pathway. These pathways are identified by differential requirement for IKK subunits. Several secondary metabolites of natural origin as well as small synthetic molecules target multiple signaling pathways including NF-ΚB-and p53. A number of natural constituents present in fruits, vegetables, and natural supplements could help in reducing or preventing chronic inflammation and related diseases.
References
- Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, et al. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci U S A 2004;101:5634-9.
- Cotran RS, Kumar V, Collins T, Robbins SL. Robbins Pathologic Basis of Disease. Philadelphia, PA: Saunders; 1999.
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140:883-99.
- Venuraju SM, Yerramasu A, Corder R, Lahiri A. Osteoprotegerin as a predioctr of coronary artery disease and cardiovascular mortality and morbidity. JACC 2010;55:2049-61.
- Lieb W, Gona P, Larson MG, Massaro JM, Lipinska I, Keaney JF Jr., et al. Biomarkers of the osteoprotegerin pathway: Clinical correlates, subclinical disease, incident cardiovascular disease, and mortality. Arterioscler Thromb Vasc Biol 2010;30:1849-54.
- Indira M, Abhilash PA. Role of NF-Kappa B (NF-ΚB) in diabetes. For Immunopathol Dis Therap 2013;4:111-32.
- Finch C, Guarascio A, Ray S, Self T. The clinical and economic burden of chronic obstructive pulmonary disease in the USA. Clinicoecon Outcomes Res 2013;5:235-45.
- Garcia-Aymerich J, Serra Pons I, Mannino DM, Maas AK, Miller DP, Davis KJ, et al. Lung function impairment, COPD hospitalisations and subsequent mortality. Thorax 2011;66:585-90.
- Miklowitz DJ, Portnoff L, Armstrong C, Keenan-Miller D, Breen C, Keely A. Inflammatory cytokines and nuclear factor-kappa B activation in adolescents with bipolar and major depression. Psychiatry Res 2016;241:315-22.
- Takahashi A, Flanigan ME, McEwen BS, Russo SJ. Aggression, social stress, and the immune system in humans and animal models. Front Behav Neurosci 2018;12:56.
- Lin TH, Pajarinen J, Lu L, Nabeshima A, Cordova LA, Yao Z, et al. NF-ΚB as a therapeutic target in inflammatory-associated bone diseases. Adv Protein Chem Struct Biol 2017;107:117-54.
- Kraus S, Arber N. Inflammation and colorectal cancer. Curr Opin Pharmacol 2009;9:405-10.
- Park MH, Hong JT. Roles of NF-kB in cancer and inflammatory diseases and their therapeutic approaches. Cells 2016;5:15.
- Li Q, Withoff S, Verma IM. Inflammation-associated cancer: NF-kappaB is the lynchpin. Trends Immunol 2005;26:318-25.
- Vlahopoulos SA, Cen O, Hengen N, Agan J, Moschovi M, Critselis E, et al. Dynamic aberrant NF-ΚB spurs tumorigenesis: A new model encompassing the microenvironment. Cytokine Growth Factor Rev 2015;26:389-403.
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 2009;1:a001651.
- Pal S, Bhattacharjee A, Ali A, Mandal NC, Mandal SC, Pal M, et al. Chronic inflammation and cancer: Potential chemoprevention through nuclear factor kappa B and p53 mutual antagonism. J Inflamm (Lond) 2014;11:23.
- Yao J, Duan L, Fan M, Wu W. Gamma-secretase inhibitors exert antitumor activity via down-regulation of Notch and Nuclear factor Kappa B in human tongue carcinoma cells. Oral Dis 2007;13:555-63.
- Chakravarti N, Kadara H, Yoon DJ, Shay JW, Myers JN, Lotan D, et al. Differential inhibition of protein translation machinery by curcumin in normal, immortalized, and malignant oral epithelial cells. Cancer Prev Res (Phila) 2010;3:331-8.
- Aggarwal S, Takada Y, Singh S, Myers JN, Aggarwal BB. Inhibition of growth and survival of human head and neck squamous cell carcinoma cells by curcumin via modulation of nuclear factor-kappaB signaling. Int J Cancer 2004;111:679-92.
- Chang KW, Hung PS, Lin IY, Hou CP, Chen LK, Tsai YM, et al. Curcumin upregulates insulin-like growth factor binding protein-5 (IGFBP-5) and C/EBPalpha during oral cancer suppression. Int J Cancer 2010;127:9-20.
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev 2004;18:2195-224.
- Karin M, Greten FR. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005;5:749-59.
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol 2007;8:49-62.
- Scheidereit C. IkB kinase complexes: Gateways to NFkB activation and transcription. Oncogene 2006;25:6685-705.
- Nam NH. Naturally occurring NF-kappa B inhibitors. Meni Rev Med Chem 2006;6:945-51.
- Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, et al. Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a Quinone methide triterpenoid. Biochem Pharmacol 2006;72:1311-21.
- Lee SJ, Bai SK, Lee KS, Namkoong S, Na HJ, Ha KS, et al. Astaxanthin inhibits nitric oxide production and inflammatory gene expression by suppressing IkB kinasedependent NF-kB activation. Mol Cells 2003;16:97-105.
- Suzuki Y, Ohgami K, Shiratori K, Jin XH, Ilieva I, Koyama Y, et al. Suppressive effects of astaxanthin against rat endotoxin-induced uveitis by inhibiting the NF-kappaB signaling pathway. Exp Eye Res 2006;82:275-81.
- Bai SK, Lee SJ, Na HJ, Ha KS, Han JA, Lee H, et al. Beta-carotene inhibits inflammatory gene expression in lipopolysaccharide-stimulated macrophages by suppressing redox-based NF-kappaB activation. Exp Mol Med 2005;37:323-34.
- Patel PS, Varney ML, Dave BJ, Singh RK. Regulation of constitutive and induced NF-kappaB activation in malignant melanoma cells by capsaicin modulates interleukin-8 production and cell proliferation. J Interferon Cytokine Res 2002;22:427-35.
- Kim CS, Kawada T, Kim BS, Han IS, Choe SY, Kurata T, et al. Capsaicin exhibits anti-inflammatory property by inhibiting ikB-a degradation in LPS-stimulated peritoneal macrophages. Cell Signal 2003;15:299-306.
- Anand P, Sung B, Kunnumakkara AB, Rajasekharan KN, Aggarwal BB. Suppression of pro-inflammatory and proliferative pathways by diferuloylmethane (curcumin) and its analogues dibenzoylmethane, dibenzoylpropane, and dibenzylideneacetone: Role of Michael acceptors and Michael donors. Biochem Pharmacol 2011;82:1901-9.
- Gupta SC, Prasad S, Kim JH, Patchva S, Webb LJ, Priyadarsini IK, et al. Multitargeting by curcumin as revealed bimolecular interaction studies. Nat Prod Rep 2011;28:1937-55.
- Harikumar KB, Kunnumakkara AB, Ahn KS, Anand P, Krishnan S, Guha S, et al. Modification of the cysteine residues in ikappaBalpha kinase and NF-kappaB (p65) by xanthohumol leads to suppression of NF-kappaB-regulated gene products and potentiation of apoptosis in leukemia cells. Blood 2009;113:2003-13.
- Shehzad A, Lee YS. Molecular mechanisms of curcumin action: Signal transduction. Biofactors 2013;39:27-36.
- Lu C, Guo Y, Yan J, Luo Z, Luo HB, Yan M, et al. Design, synthesis, and evaluation of multitarget-directed resveratrol derivatives for the treatment of alzheimer's disease. J Med Chem 2013;56:5843-59.
- Michan S, Sinclair D. Sirtuins in mammals: Insights into their biological function. Biochem J 2007;404:1-3.
- Saunders LR, Verdin E. Sirtuins: Critical regulators at the crossroads between cancer and aging. Oncogene 2007;26:5489-504.
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 2004;23:2369-80.
- Nicholas C, Batra S, Vargo MA, Voss OH, Gavrilin MA, Wewers MD, et al. Apigenin blocks lipopolysaccharide-induced lethality in vivo and proinflammatory cytokines expression by inactivating NF-kappaB through the suppression of p65 phosphorylation. J Immunol 2007;179:7121-7.
- Xu L, Zhang L, Bertucci AM, Pope RM, Datta SK. Apigenin, a dietary flavonoid, sensitizes human T cells for activation-induced cell death by inhibiting PKB/Akt and NF-kappaB activation pathway. Immunol Lett 2008;121:74-83.
- Natarajan K, Manna SK, Chaturvedi MM, Aggarwal BB. Protein tyrosine kinase inhibitors block tumor necrosis factor-induced activation of nuclear factor-kappaB, degradation of ikappaBalpha, nuclear translocation of p65, and subsequent gene expression. Arch Biochem Biophys 1998;352:59-70.
- Sarkar FH, Li Y. Mechanisms of cancer chemoprevention by soy isoflavone genistein. Cancer Metastasis Rev 2002;21:265-80.
- Ju W, Wang X, Shi H, Chen W, Belinsky SA, Lin Y, et al. A critical role of luteolin-induced reactive oxygen species in blockage of tumor necrosis factor-activated nuclear factor-kappaB pathway and sensitization of apoptosis in lung cancer cells. Mol Pharmacol 2007;71:1381-8.
- Mackenzie GG, Oteiza PI. Modulation of transcription factor NF-kappa B in Hodgkin's lymphoma cell lines: Effect of (-)-epicatechin. Free Radic Res 2006;40:1086-94.
- Joo S, Song Y, Park Y, Myung E, Chung C, Park KJ, et al. Epigallocatechin gallate inhibits phorbol ester-induced activation of NF-kappa B and CREB in mouse skin: Role of p38 MAPK. Ann N Y Acad Sci 2007;1095:504-12.
- Wheeler DS, Catravas JD, Odoms K, Denenberg A, Malhotra V, Wong HR. Epigallocatechin-3-gallate, a green tea-derived polyphenol, inhibits IL-1 beta-dependent proinflammatory signal transduction in cultured respiratory epithelial cells. J Nutr 2004;134:1039-44.
- Kundu JK, Surh YJ. Epigallocatechin gallate inhibits phorbol ester-induced activation of NF-kappa B and CREB in mouse skin: Role of p38 MAPK. Ann N Y Acad Sci 2007;1095:504-12.
- Cheon S, Chung K, Jeon E, Nuhroho A, Park H. Anti-inflammatory activity of saxifragin via inhibition of NF-kB involves capsase-1 activation. J Nat Prod 2015;78:1579-85.
- Harikumar KB, Sung B, Tharakan ST, Pandey MK, Joy B, Guha S, et al. Sesamin manifests chemopreventive effects through the suppression of NF-kappa B-regulated cell survival, proliferation, invasion, and angiogenic gene products. Mol Cancer Res 2010;8:751-61.
- Jeng KC, Hou RC, Wang JC, Ping LI. Sesamin inhibits lipopolysaccharide-induced cytokine production by suppression of p38 mitogen-activated protein kinase and nuclear factor-kappaB. Immunol Lett 2005;97:101-6.
- Gao X, Deeb D, Liu Y, Gautam S, Dulchavsky SA, Gautam SC, et al. Immunomodulatory activity of xanthohumol: Inhibition of T cell proliferation, cell-mediated cytotoxicity and th1 cytokine production through suppression of NF-kappaB. Immunopharmacol Immunotoxicol 2009;31:477-84.
- Medeiros R, Passos GF, Vitor CE, Koepp J, Mazzuco TL, Pianowski LF, et al. Effect of two active compounds obtained from the essential oil of Cordia verbenacea on the acute inflammatory responses elicited by LPS in the rat paw. Br J Pharmacol 2007;151:618-27.
- Takada Y, Murakami A, Aggarwal BB. Zerumbone abolishes NF-kB and IkBa kinase activation leading to suppression of antiapoptotic and metastatic gene expression, upregulation of apoptosis, and downregulation of invasion. Oncogene 2005;24:6957-69.
- Murakami A, Matsumoto K, Koshimizu K, Ohigashi H. Effects of selected food factors with chemo preventive properties on combined lipopolysaccharide-and interferon-g-induced IkB degradation in RAW264.7 macrophages. Cancer Lett 2003;195:17-25.
- Lo AH, Liang YC, Lin-Shiau SY, Ho CT, Lin JK. Carnosol, an antioxidant in rosemary, suppresses inducible nitric oxide synthase through down-regulating nuclear factor-kB in mouse macrophages. Carcinogenesis 2002;23:983-91.
- Huang SC, Ho CT, Lin-Shiau SY, Lin JK. Carnosol inhibits the invasion of B16/F10 mouse melanoma cells by suppressing metalloproteinase-9 through down regulating nuclear factor-kB and c-Jun. Biochem Pharmacol 2005;69:221-32.
- Wang ZF, Tang XC. Huperzine A protects C6 rat glioma cells against oxygen-glucose deprivation-induced injury. FEBS Lett 2007;581:596-602.
- Koo TH, Lee JH, Park YJ, Hong YS, Ki HS, Kim KW, et al. A sesquiterpene lactone, costunolide, from Magnolia grandiflora inhibits NF-kB by targeting IkB phosphorylation. Planta Med 2001;67:103-7.
- Han JW, Lee BG, Kim YK, Yoon JW, Jin HK, Hong S, et al. Ergolide, sesquiterpene lactone from Inula britannica, inhibits inducible nitric oxide synthase and cyclo-oxygenase-2 expression in RAW264.7 macrophages through the inactivation of NF-kB. Br J Pharmacol 2001;133:503-12.
- Lyss G, Knorre A, Schmidt TJ, Pahl HL, Merfort I. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-kB by directly targeting p65. J Biol Chem 1988;273:33508-16.
- Wang CN, Shiao YJ, Lin YL, Chen CF. Nepalolide A inhibits the expression of inducible nitric oxide synthase by modulating the degradation of IkB-a and IkB-b in C6 glioma cells and rat primary astrocytes. Br J Pharmacol 1999;128:345-56.
- Kwok BH, Koh B, Ndubuisi MI, Elofsson M, Crews CM. The anti-inflammatory natural product parthenolide from the medicinal herb feverfew directly binds to and inhibits ikappaB kinase. Chem Biol 2001;8:759-66.
- Ischikawa H, Nair MS, Takada Y, Sheeja DB, Kumar MA, Oommen OV, et al. Isodeoxyelephantopin, a novel sesquiterpene lactone, potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis through suppression of nuclear factor-kB (NF-kB) activation and NF-kB-regulated gene expression. Clin Cancer Res 2006;12:5910-8.
- Ojo-Amaize EA, Kapahi P, Kakkanaiah VN, Takahashi T. Shalom-Barak T, Cottam HB, et al. Hypoestoxide, a novel anti-inflammatory natural diterpene, inhibits the activity of IkB kinase. Cell Immunol 2001;209:149-57.
- Ojo-Amaize EA, Cottam HB, Oyemade OA, Okogun JI, Nchekwube, EJ. Hypoestoxide inhibits tumor growth in the mouse CT26 colon tumor model. World J Gastroenterol 2007;13:4586-8.
- Koo HJ, Song YS, Kim HJ, Lee YH, Hong SM, Kim SJ, et al. Antiinflammatory effects of genipin, an active principle of gardenia. Eur J Pharmacol 2004;495:201-8.
- Jeong HJ, Koo HN, Na HJ, Kim MS, Hong SH, Eom JW, et al. Inhibition of TNF-a and IL-6 production by aucubin through blockade of NF-kB activation in RBL-2H3 mast cells. Cytokine 2002;18:252-9.
- Chang IM. Liver-protective activities of aucubin derived from traditional oriental medicine. Res Commun Mol Pathol Pharmacol 1998;102:189-204.
- Kang HS, Kim YH, Lee CS, Lee JJ, Choi I, Pyun KH. Suppression of interleukin-1 and tumor necrosis factor-a production by acanthoic acid, (-)-pimara- 9(11),15-dien-19-oic acid, and it antifibrotic effects in vivo. Cell Immunol 1996;170:212-21.
- Kim JY, Jung KS, Lee KJ, Na HK, Chun HK, Kho YH, et al. The coffee diterpene kahweol suppress the inducible nitric oxide synthase expression in macrophages. Cancer Lett 2004;213:147-54.
- Kim JY, Jung KS, Jeong HG. Suppressive effects of the kahweol and cafestol on cyclooxygenase-2 expression in macrophages. FEBS Lett 2004;569:321-6.
- Kim SW, Choi SC, Choi EY, Kim KS, Oh JM, Lee HJ, et al. Catalposide, a compound isolated from Catalpa ovata, attenuates induction of intestinal epithelial proinflammatory gene expression and reduces the severity of trinitrobenzene sulfonic acid-induced colitis in mice. Inflamm Bowel Dis 2004;10:564-72.
- Jang SI, Kim HJ, Kim YJ, Jeong SI, You YO. Tanshinone IIA inhibits LPS-induced NF-kappaB activation in RAW 264.7 cells: Possible involvement of the NIK-IKK, ERK1/2, p38 and JNK pathways. Eur J Pharmacol 2006;542:1-7.
- Liu H, Liu ZH, Chen ZH, Yang JW, Li LS. Triptolide: A potent inhibitor of NF-kappa B in T-lymphocytes. Acta Pharmacol Sin 2000;21:782-6.
- Garcia PA, de Oliveira AB, Batista R. Occurrence, biological activities and synthesis of kaurane diterpenes and their glycosides. Molecules 2007;12:455-83.
- Takada Y, Aggarwal BB. Betulinic acid suppresses carcinogen-induced NF-kB activation through inhibition of IkBa kinase and p65 phosphorylation: Abrogation of cyclooxygenase-2 and matrix metalloprotease-9. J Immunol 2003;171:3278-86.
- Cherng JM, Lin HJ, Hung M, Lin YR, Chan MH, Lin JC. Inhibition of nuclear factor kB is associated with neuroprotective effects of glycyrrhizic acid on glutamate-induced excitotoxicity in primary neurons. Eur J Pharmacol 2006;547:10-21.
- Myers JN, Altevogt P, Yuen AP. Lupeol suppresses cisplatin-induced nuclear factor-kB activation in head and neck squamous cell carcinoma and inhibits local invasion and nodal metastasis in an orthotopic nude mouse model. Cancer Res 2007;67:8800-9.
- Fernandez MA, de las Heras B, Garcia MD, Saenz MT, Villar A. New insights into the mechanism of action of the anti-inflammatory triterpene lupeol. J Pharm Pharmacol 2001;53:1533-9.
- Saleem M, Afaq F, Adhami VM, Mukhtar H. Lupeol modulates NF-kappaB and PI3K/Akt pathways and inhibits skin cancer in CD-1 mice. Oncogene 2004;23:5203-14.
- Wang H, Syrovets T, Kess D, Buchele B, Hainzl H, Lunov O, et al. Targeting NF-kappa B with a natural triterpenoid alleviates skin inflammation in a mouse model of psoriasis. J Immunol 2009;183:4755-63.
- Dai Y, Desano J, Tang W, Meng X, Meng Y, Burstein E, et al. Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and NF-kappaB. PLoS One 2010;5:e14153.
- Shishodia S, Majumdar S, Banerjee S, Aggarwal BB. Ursolic acid inhibits nuclear factor-kB activation induced by carcinogenic agents through suppression of IkBa kinase and p65 phosphorylation: Correlation with down regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1.Cancer Res 2003;63:4375-83.
- Harikumar KB, Sung B, Pandey MK, Guha S, Krishnan S, Aggarwal BB, et al. Escin, a pentacyclic triterpene, chemosensitizes human tumor cells through inhibition of nuclear factor-kappaB signaling pathway. Mol Pharmacol 2010;77:818-27.
- Leung CY, Liu L, Wong RN, Zeng YY, Li M, Zhou H. Saikosaponin-d inhibits T cell activation through the modulation of PKC, JNK, and NF-kB transcription factor. Biochem Biophys Res Commun 2005;338:1920-7.
- Haridas V, Arntzen CJ, Gutterman JU. Avicins, a family of triterpenoid saponins from Acacia victoriae (Bentham), inhibit activation of nuclear factor-kB by inhibiting both its nuclear localization and ability to bind DNA. Proc Natl Acad Sci USA 2001;98:11557-62.
- Choi K, Kim M, Ryu J, Choi C. Ginsenosides compound K and Rh2 inhibit tumor necrosis factor-a-induced activation of the NF-kB and JNK pathways in human astroglial cells. Neurosci Lett 2007;421:37-41.
- Zhang J, Cao L, Wang H, Cheng X, Wang L, Zhu L, et al. Ginsenosides regulate PXR/NF-ΚB signaling and attenuate dextran sulfate sodium-induced colitis. Drug Metab Dispos 2015;43:1181-9.
- Li J, Zhong W, Wang W, Hu S, Yuan J, Zhang B, et al. Ginsenoside metabolite compound K promotes recovery of dextran sulfate sodium-induced colitis and inhibits inflammatory responses by suppressing NF-ΚB activation. PLoS One. 2014;9:e87810.
- Karunaweera N, Raju R, Gyengesi E, Munch G. Plant polyphenols as inhibitors of NE-kB induced cytokine production-a potential anti-inflammatory treatment for Alzheiemr's disease. Front Mol Neurosci 2015;8:24.
- Calixto JB, Campos MM, Otuki MF, Santos AR. Anti-inflammatory compounds of plant origin. Part II. Modulation of pro-inflammatory cytokines, chemokines and adhesion molecules. Planta Med 2004;70:93-103.